




吻合器的前世今生
2020-07-31
医学的发展随着人类科学技术的发展而不断进步,外科手术从最初鲜血淋漓、九死一生的状况进步到现在的“微创/无创、无痛、安全、快速、有效”绝非一人或一日之功。在外科从蒙昧走向科学、文明的近两百年历史中,手术器械的发明和改进无疑发挥了至关重要的作用。在现代科学快速发展的背景下,可以说很多先进科技都会最先应用于医学领域,外科手术器械更新迭代加速,带来更多利好和可能。
缝合是外科手术中必不可少且非常重要的环节。二十世纪前,外科手术中所有的缝合工作都靠手工缝线完成,遇到小伤口或小血管的结扎还好,一旦遇到食管、肺、胃肠、肝脾胰这类内脏器官的手术,医生就得在缝合上花费几个甚至十几个小时。如今很多手术依然需要医生运用高超的“缝纫”技巧对伤口或器官组织进行缝合。但有时他们也能够借助一种医学“订书机”——吻合器,通过机械化的操作方式让手术缝合更加高效,并让许多困难复杂的手术变得简便,且大幅降低了手术并发症发生率。

1、1908年,匈牙利医生Hümer Hültl从订书机上获得灵感,成功发明出一种新的止血缝合工具——吻合器
它由各种金属部件组装而成,重达8磅,装配费时达2个小时。看它的中文名字你可能没有什么感觉,可如果搬出它的英文名称Surgical Stapler(外科订书机),是不是就形象很多?

“外科缝合器之父”——Humer Hultl↑↑↑
既然是从订书机上获得的灵感,想必大家立刻就能猜到吻合器的工作原理,回忆一下平时你装订纸张的过程,把纸替换成我们体内的组织,就能大概还原吻合器工作时的样子了。
当然,如果和文具订书机完全一样,吻合器就称不上是一件伟大的发明了。为了更好解决手术中的止血问题,吻合器的发明者Hültl医生颇有一些很棒的想法:
他改造了钉砧,让受到挤压后的缝钉能呈B字型,这样既能牢牢扎住主要 血管,又能保证钉合组织和切割边缘的血供和营养;他设计的多排缝钉采用交错排列法,缝钉的轨迹如两条平行的虚线,彼此锁住空隙,确保切割所经的所有血管都被结扎,以避免吻合处有渗出现象。
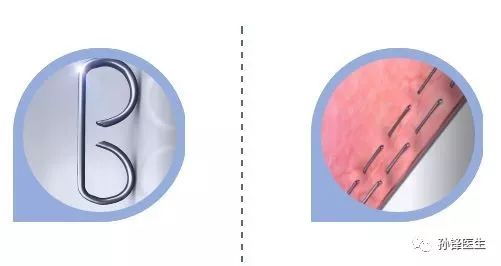
“B”字型缝钉↑↑↑
值得一提的是,直到今天这两点还被应用于吻合器的设计中。因此Hultl被誉为“外科缝合器之父”。
吻合器的出现大大提升了一些手术的效率和成功率,而它本身也经历了不断的升级换代:原本的吻合器只能缝合,之后人们在钉仓中加入刀片,实现了边切边缝的功能;应用于不同器官手术的吻合器,在外形和功能上也发生了不同的演变。
2、1921年,更轻便的现代吻合器登场
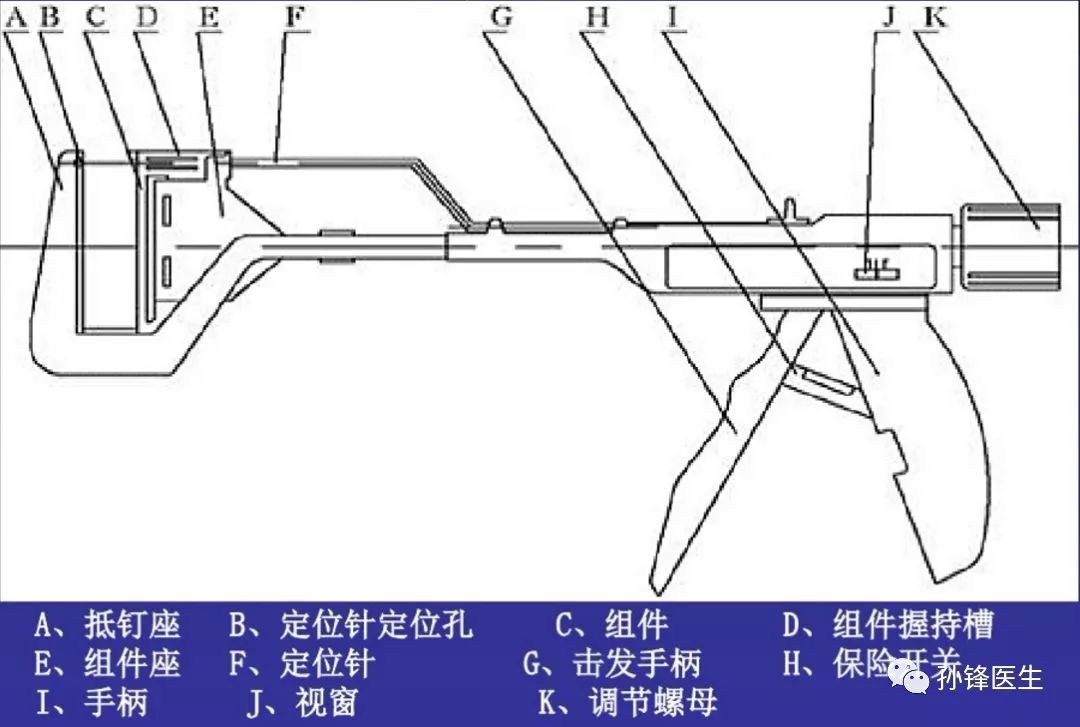
直线型缝合器基本结构(图片来源:网络)↑↑↑
1921年直线型缝合器问世 (Aladar Von Petz, 匈牙利),简化了吻合器的设计,用镍银合金代替金属丝,并且可以重复填装缝钉,使得吻合器更加轻便。1934年德国的H. Friederich和Neuffer对缝合器作了改进,加装了可更换钉仓;1951年前苏联的实验外科器械研究所对缝合器进行了系统的研究,并在此领域处于领先水平。
3、1958年,吻合器在美国获得关键性改良
1958年美国学者Ravitch在前苏联实验外科器械研究所参观后,将缝合器技术引进美国。
4、1967年,临床手术吻合器的发展
这一年,美国外科公司Auto Suture的创始人Locn Hirsch以及他的工程师们,从根本上解决了装配缝钉费时的问题,生产出一种可以方便应用于临床手术的吻合器。
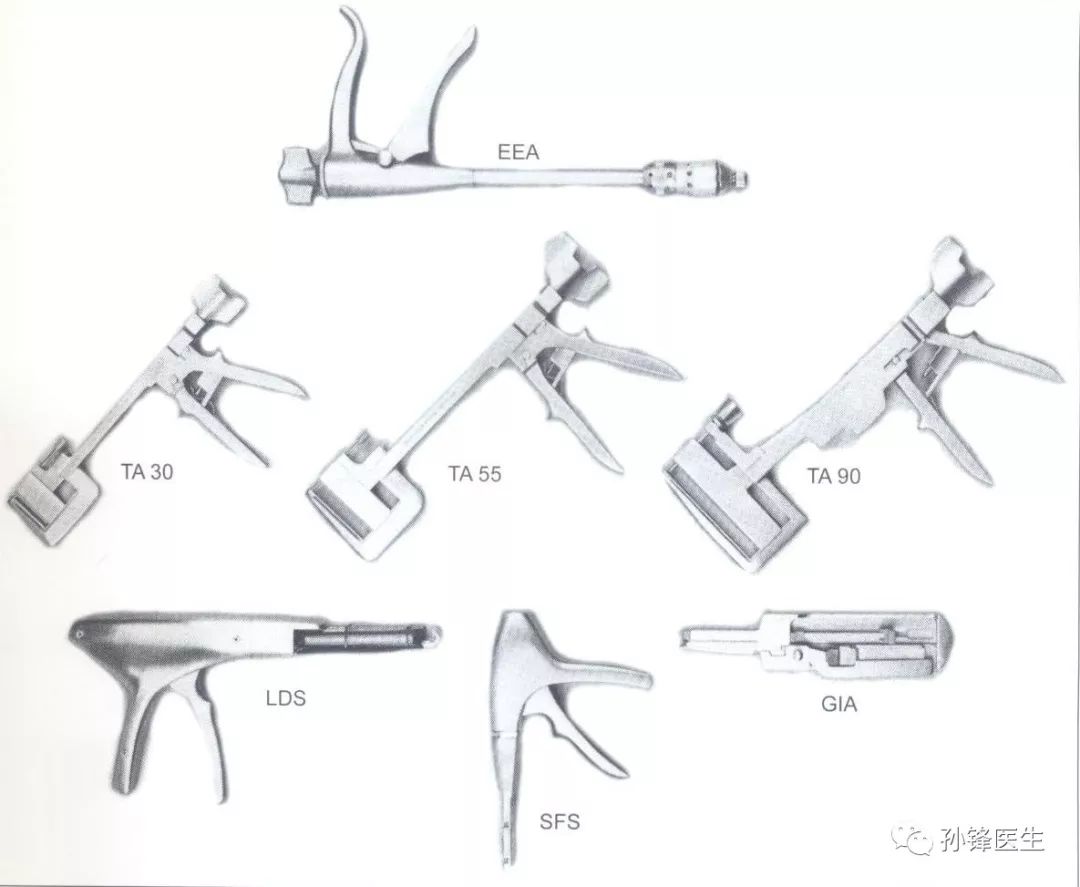
发展中的吻合器↑↑↑
5、1968年,切割缝合器问世
美国外科公司推出具有双组双排缝钉及刀片的吻合器,在缝合的同时可以进行组织切割,多用于胃肠组织的离断,或胃肠、肠肠的内翻吻合。
6、1978年,美国外科公司发明了管型端端吻合器(EEA)
EEA具有双排环形缝钉及刀片,刀片用于吻合时切断缝钉内侧的组织,使之形成端端吻合口,用于不同直径腔道的环状吻合)。
管型端端吻合器的出现↑↑↑
7、1979年,一次性吻合器问世,吻合器应用开始普及化、细分化
1979年美国Ethicon公司,研制和发展了多种缝合器和吻合器,并生产出全球第一把完全一次性、单病人使用的机械吻合器。一次性吻合器减少了器械消毒带来的患者交叉感染的几率,降低了患者术后并发症发生的概率,也使吻合器的大批生产和广泛应用又上了一个台阶。
此后,各类用于不同组织的缝合器陆续问世,为外科医生们提供了手术用的有力武器。

一次性吻合器的问世↑↑↑
8、缝钉成型的技术也在不断地改良创新
2004年,Covidien公司发明了DST缝钉成型技术。DST是Directional Stapling Technology的缩写,翻译过来是“导向型缝钉成型技术”。和传统技术相比,DST技术做了两点革新:
(1)近似方柱形钉腿:横截面近似矩形的方柱形缝钉与推钉板接触的是一个平面,避免推钉板行进时,钉腿发生偏移或扭曲。

近似方柱形钉腿↑↑↑
(2)水滴状钉砧:面积更大的钉砧凹槽可以更有效地引导钉腿完美成形。

水滴状钉砧↑↑↑
这两点共同作用的结果,就是无论需要吻合的组织是厚是薄,都能稳定压出一个处女座也挑不出错的完美B字缝钉。
9、2012年,Covidien公司发明了Tristaple创世智能吻合技术
传统吻合器工作时,会在切割路径两边各击发2至3排等高、等尺寸的缝钉。但实际应用时,由于组织厚度存在差异,等高的缝钉难免“顾此失彼”,钉得紧,可能会影响血供和愈合 ;钉得松,可能产生渗漏,真是左右为难。
为了解决这对矛盾,美敦力推出了Tri-Staple™ 技术,简单说来,就是采用了三排不等高的、呈阶梯状的缝钉技术。
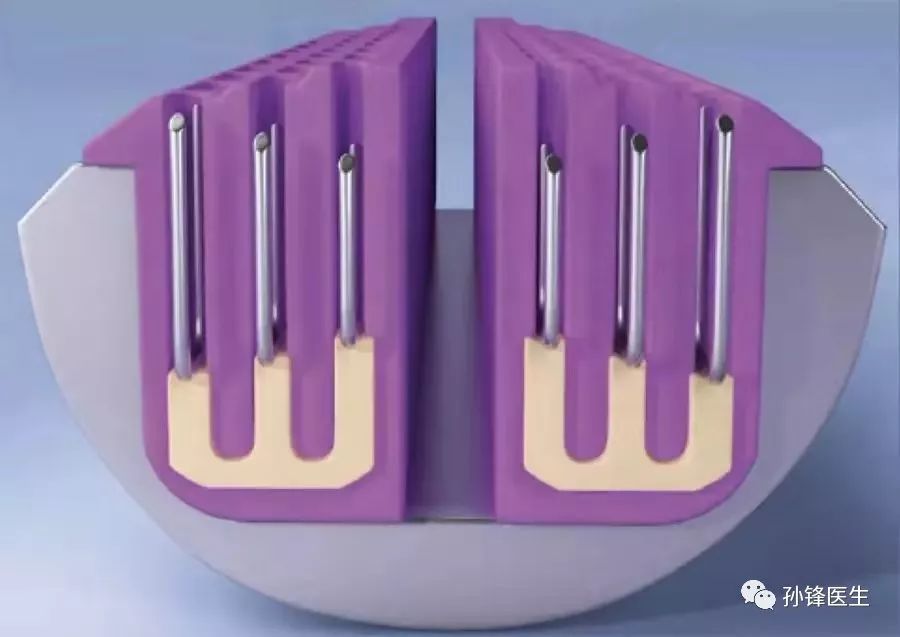
Tri-Staple™ 技术↑↑↑
对缝钉高度的简单调整,带来的却是不简单的效果。击发后的缝钉内紧外松,靠近刀口的钉腿短,缝得较紧,能防止出血;远端钉腿高,缝得较松,能维持组织液和血液的流通,为组织提供营养,加快愈合。
此外,缝钉的击发时间也有先后,从内侧到外侧三排依次击发,配合阶梯型的钉匣面,夹得又准又不伤害组织。同时,在击发过程中,缝钉始终先于刀片,这样的“先缝后切”也最大程度确保了组织的安全。

“先缝后切”技术↑↑↑
总之,Tri-Staple技术通过三排不等高和渐进性夹闭的独特设计,实现了吻合严密、适配组织厚度和预防损伤的效果,大大提升了患者的术中安全和术后愈合。
10、2015年,世界上第一个电动智能切割吻合器诞生

电动智能切割吻合器↑↑↑
Covidie公司在2015年推出了iDrive™智动平台,机械吻合需要医生不断操纵手柄分段击发缝钉,如果击发速度不当,或是操作时发生移位,都会对患者的组织造成伤害,安全性得不到保障。而iDrive智动平台不仅能自动调节击发速度,匀速击发,还能在夹取组织厚度过厚时给予反馈,提醒医生改变位置或更换另一种高度的钉腿,有效提高了吻合过程中组织的安全性。更不用提只需简单操控手柄处的按钮就能实现钉仓角度的无极旋转,大大方便了医生操作。
来源:孙锋医生


