




多家械企开始布局宠物器械:迈瑞、万孚…
2021-11-02
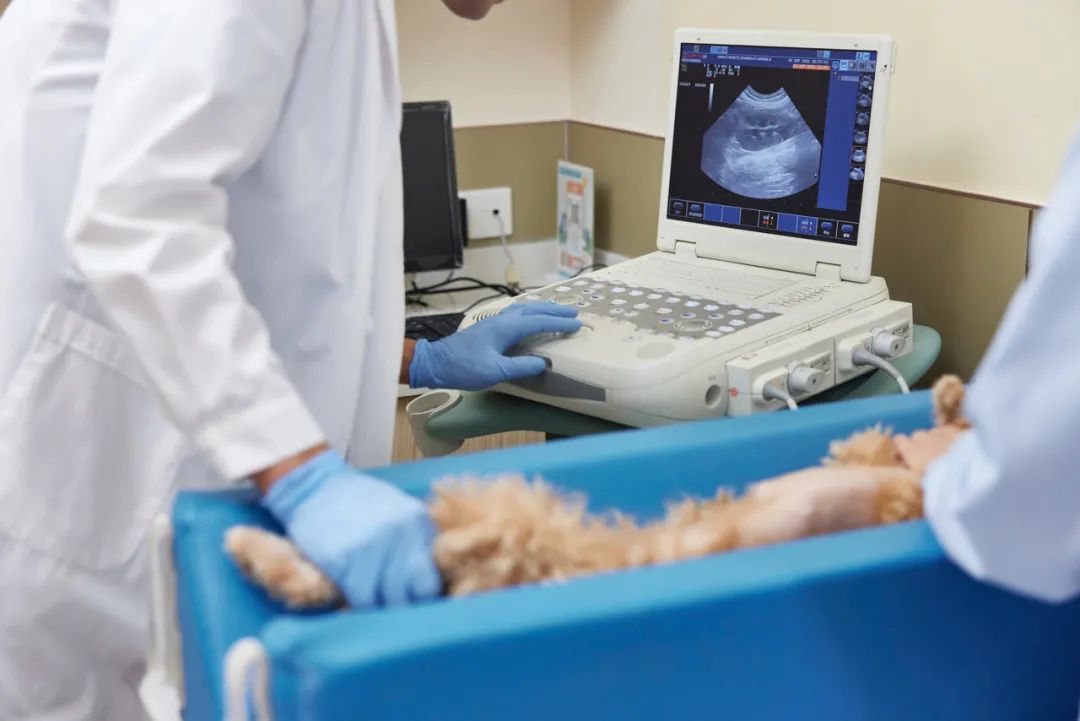
宠物医疗需要更强的专业化,比如动物的白内障手术、骨头矫正等等。而一些核磁、洗牙机、血液分析仪、呼吸机、麻醉机等也成为宠物医院的配置刚需。
兽用医疗器械采购量持续攀升
10月28日,是第12个中国兽医日,伴随公共卫生事业的进程,兽医的治疗范畴已经扩大到人畜共患疾病的诊疗,也波及到医药工业等多个领域,其中影响最大的是宠物医疗市场快速崛起,不仅带动了从业人员的增加,也使消费模式发生了根本性变化。
有投资人指出:消费市场的增量空间排序是女性、儿童、老人、宠物、男性。
天猫曾披露双十一数据,宠物奶粉的销量已经超过婴幼儿奶粉。而在医疗器械领域,宠物医疗潜力更是不容小觑。根据前瞻产业研究院发布的《2020-2025年中国宠物行业市场前景预测与投资战略规划分析报告》分析,未来我国宠物医疗市场规模达到1000亿元。
10月24日,迈瑞医疗公布消息,目前迈瑞动物医疗已经正式入驻新总部,业务上主要覆盖超声、体外诊断和生命信息与支持等产品;生产基地分为3个区域,8条生产线,涵盖血液分析仪光学装配、超声影像设备生产、呼吸麻醉机生产。

广州万孚生物旗下的全资子公司万德康,是宠物医疗器械制造方面比较有代表性的企业,有专用于动物的免疫荧光平台、动物即时凝血平台、干式生化平台等。
目前任职于宠物医院的设备采购处的陈树斌告诉赛柏蓝器械:国内宠物医疗的市场要远比人们想象中的要大很多。以深圳为例,平均每三个家庭中就有一家养宠物,而宠物行业的消费总额也非常高,且大多数一线二线城市的宠物主比较倾向于给宠物买高昂的日用品。
“大多数的宠物主,在健康检查和治疗上倾向于高端的全流程的服务,大型宠物医院配备的科室也比较齐全,神经外科、软组织科、眼科、牙科、皮肤科、影像科等客流量很高,有的医院还配有肿瘤科”
陈树斌指出,目前宠物主最大的需求主要是宠物疫苗、宠物美容、宠物饮食和宠物体检。而宠物疫苗注射是目前营收中最大的一块。
“在费用上,宠物体检也是消费量最大的一块之一,里面主要涉及了动物免疫、预防。医院在检验检测试剂、诊疗仪、超声影像系统、尿液分析仪、生化仪方面有较强的采购量和消耗量,且这一部分国产与进口的品牌在价格上差异还是比较明显的,国内企业的销售价格也比较低,性价比也弱一点,但利润也会高一些。”
高端设备检查是竞争力所在
国内宠物数量的增加,给宠物各个子行业带来了极大的市场刚需,但我国的市场与发达国家相比还是有比较大的增量空间。2010年之后,国内宠物市场发展进入到了最快的增长期,多地发布关于宠物犬业发展规划。
赛柏蓝器械梳理得知,文件主要强调了要推进宠物饲料生产的企业的扶持、增加外销出口的比例等,在市场准许方面加以扶持。
从2010年前后,宠物市场也开始形成了连锁服务、网络营销、宠物训练、宠物美容以及宠物云平台等全产业链的完善,加盟店、旗舰点等经营模式逐步闯进视野。
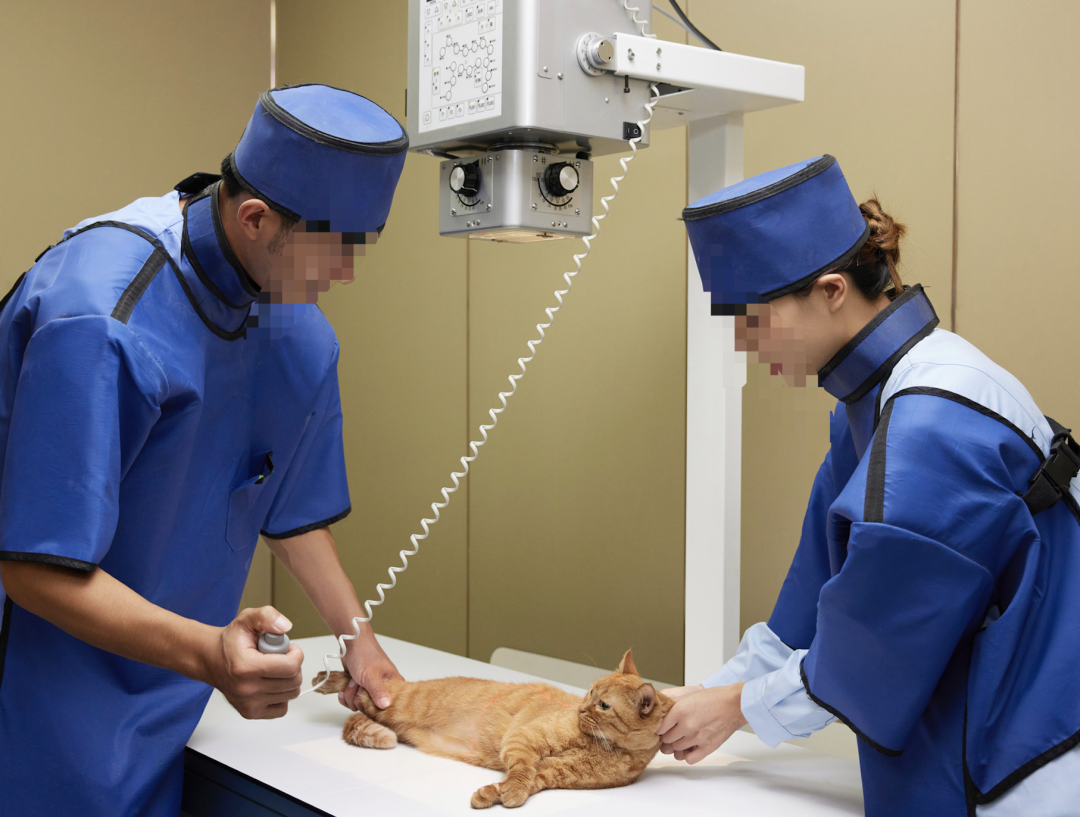
陈树斌指出,目前种中国的宠物市场,行业产业链已经分为了上游、中游、下游三个部分,而在医疗领域,主要分为耗材采购、药品采购、设备购买以及第三方检测。
在上游领域,新华医疗、迈瑞医疗、奥林巴斯等等都有相关的产业涉及;而在药品方面,金盾药业、瑞普生物、拜耳等也都是热门的采购企业。
中游就是一些全国连锁的动物医院,比如瑞鹏宠物医院等等,已经形成了多家连锁的接机构。
陈树斌特别强调,动物保险方面也是一个比较热门的兴趣子行业,中国平安、太平洋保险等等都有相关的险种。
宠物店郭丽珍对赛柏蓝器械解释到,很多宠物主也比较倾向于连锁的机构,对于运营者也倾向于选择加盟店,这对技术、设备和培训方面收获更大,在成本控制、供应链方面也更加省时省力。
对于未来发展方向,郭丽珍指出,宠物医疗需要更强的专业化,比如动物的白内障手术、骨头矫正等等,高端设备检查一定是竞争力所在,这一类技术门槛、技术壁垒还比较高,一些宠物医院会配备一些核磁、洗牙机等,可以更好地给动物进行体检跟诊断,但一些小的宠物医院不能够所进行的。另外在电子病历管理方面,也需要亟待加强。
来源:小桔灯


