




全球心室辅助装置(VAD)领域佼佼者阿比奥梅德ABIOMED
2021-09-09
阿比奥梅德(Abiomed,NASDAQ: ABMD)是全球心室辅助血泵(VAD)领域佼佼者,位列“2020全球医疗器械100强”榜单74名。公司生产的Impella心脏泵,是唯一获得美国 FDA 批准适用于需要高危 PCI 或 AMI 心源性休克等严重冠状动脉疾病患者的经皮心脏泵技术。目前全球使用Impella心脏泵的患者超过15万。2019 年,巴伦周刊(Barron’s)将 Abiomed 列为过去十年表现第四佳的股票,总回报率达1,983%。
一、公司创始人
Abiomed成立于1981年,由David M. Lederman创立,总部位于美国马萨诸塞州丹弗斯市。
Lederman先生是一名航空航天工程师,出生于哥伦比亚波哥大,为波兰移民后裔。1966年毕业于康奈尔大学,获得工程、物理和数学学位,后又在康奈尔攻读了航空航天工程的硕士和博士学位。创办 Abiomed之前,他在一家航空航天技术公司从事心脏辅助技术的研究。创办 Abiomed的前一年,他50 多岁的父亲因心脏病去世。

(图:David M. Lederman)
Lederman创立Abiomed公司的目的,是希望开发出一个人工心脏作为移植的桥梁,帮助严重虚弱的心脏病患者延长生命,为他们在移植等待期间提供更大程度的独立性。他与公司首席科学官 Robert Kung 博士合作,组建了一个设计 AbioCor 的研究团队。
AbioCor是全球第一款完全植入式心脏替代设备,相比当时其他的人工心脏产品Jarvik-7及SynCardia有很大的不同,后两者都需要由体外压缩机空气驱动。AbioCor是一种葡萄柚大小的装置,可取代患者的心脏,使用可充电电源运行,没有电线或管子穿过皮肤。其内部电池由经皮能量传输 (TET) 系统充电,这意味着没有电线或管子穿透皮肤,从而降低了感染风险。

在 2001 年至 2004 年的临床试验期间,仅有14 台 AbioCor 设备被植入人体,其中寿命最长的接受者存活了 512 天。AbioCor体积太大,并不适应于大多数人,只适合某些骨架大的男人。尽管后来开发的AbioCor II,预计比原来尺寸小三分之一,寿命最长持续五年,但截至 2016 年 3 月,AbioCor II 尚未实现。据媒体2015年的报道,AbioMed 已放弃了对该产品的进一步开发。
2012 年 8 月,Lederman 死于胰腺癌,享年68岁。尽管 AbioCor 并未在众多患者身上使用,但它为完全独立的人工心脏技术的进一步发展铺平了道路,包括衍生出的现在非常普遍用作移植桥梁的左心室辅助装置。
二、公司发展历史
1987年,在成立6年后,Abiomed登陆纳斯达克上市,股票代码为ABMD。上市头年,公司营业收入达 150 万美元。
1992年,BVS 5000® 成为全球第一个获得FDA 批准,用于心脏切开术后患者临时循环支持的心室辅助装置。BVS 5000是一种两腔体外循环辅助装置,安全、简单、有效,循环支持的脉动比其他系统更具优势。
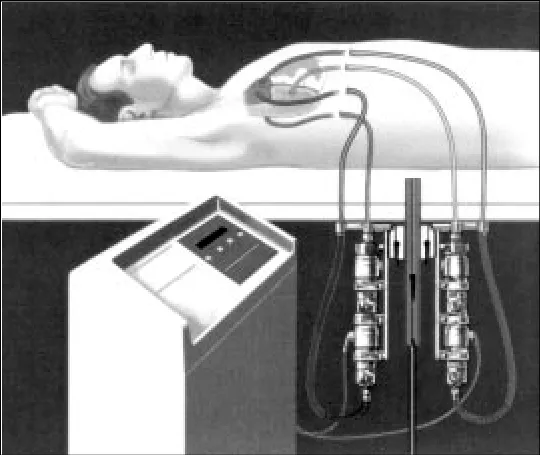
(图:BVS 5000®)
2001年7月,公司研发的完全植入式人造心脏AbioCor被医生首次植入人体。患者在植入后存活了 151 天,随后发生了致命的脑血管意外。《时代》杂志在 2001 年底授予 AbioCor 年度发明奖。
尽管经过了20多年的开发,AbioCor迟迟未能得到FDA的上市批准,甚至由于在2000年临床试验失败,导致公司股价大跌 (2000年底Abiomed的股价从年初每股41美元跌至3美元左右),濒临破产。
2004年,公司迎来新任首席执行官兼总裁 Michael R. Minogue 。据Abiomed官网介绍, Minogue任职期间,Abiomed成为增长最快、GAAP(按公认会计准则)盈利的医疗技术公司之一,过去十年中Abiomed在标普 500 指数中的股票表现排名第3位,公司目前市值为149亿美元,市盈率达96.41。

(图:公司首席执行官兼总裁 Michael R. Minogue )
Minogue曾在通用电气医疗集团(General Electric Healthcare)工作了11年,担任多个领导职位并拥有3项专利,在癌症和心血管疾病诊断成像的销售、营销、产品开发、信息技术和软件服务运营等方面积累了专业知识。他还曾在美国陆军担任军官,拥有西点军校的工程管理学学士学位和芝加哥大学MBA学位。
Minogue上任后,通过从德国初创公司Impella CardioSystems收购和开发Impella®(世界上最小的心脏泵)及体外膜肺氧合 (ECMO)等技术,将Abiomed的发展重心逐渐转向其创建的心肺康复领域。

2005年,Impella 获得 CE 标志并在欧洲上市。2008年,Impella 2.5®心脏泵获得FDA 510(k) 许可。Impella 2.5®心脏泵,流速高达每分钟2.5 L,可在高风险 PCI(经皮冠状动脉介入治疗)手术或心源性休克期间暂时维持血流。当医生治疗上述疾病时,它可以让心脏得到休息并将血液泵送到重要器官。
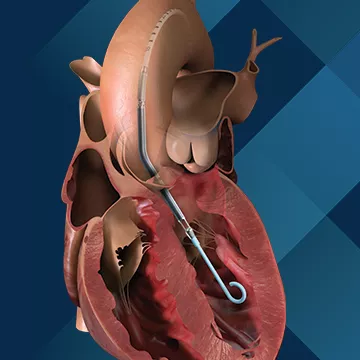
(图:Impella 2.5®)
2009年,AB便携式驱动器获 FDA 批准,首位使用 AB5000™ 的患者出院。同年,Impella 5.0®和 Impella LD®心脏泵获得FDA 510(k) 上市许可。
Impella 5.0®临时微创心脏泵,流速高达每分钟 5.0 L,已获得最长 14 天的左心室支持许可,能为危重患者提供更长持续时间的循环支持,给医生更长的时间来评估心脏康复的程度。Impella LD 是一种通过手术植入的心脏泵,可在手术期间和手术后为心脏提供临时支撑。如通过靠近肩部的腋动脉插入Impella 5.0 和 Impella LD 心脏泵,患者可以在其支撑下四处走动。
2011年,新型Impella自动控制器™ (AIC) 上市。同年,RECOVER I 临床试验结果发表在《胸心血管外科杂志》上,该实验是一项关于 Impella 5.0/LD 用于心脏术后循环支持的多中心前瞻性研究。试验结果表明,对于出现心源性休克的患者,使用 Impella 5.0/left direct 装置是安全可行的。
2012年,PROTECT II 随机对照试验结果发表在美国心脏协会杂志《循环》(Circulation)上。该试验有关使用Impella 2.5和IABP提供血动力支持的患者在接受高风险PCI手术后的效果研究。试验发现接受 Impella 2.5循环支持的高危PCI患者在术后90 天时的主要不良事件少于接受主动脉内球囊泵 (IABP)介入治疗的患者,这为后续Impella RP获得FDA批复鉴定了重要的试验基础。
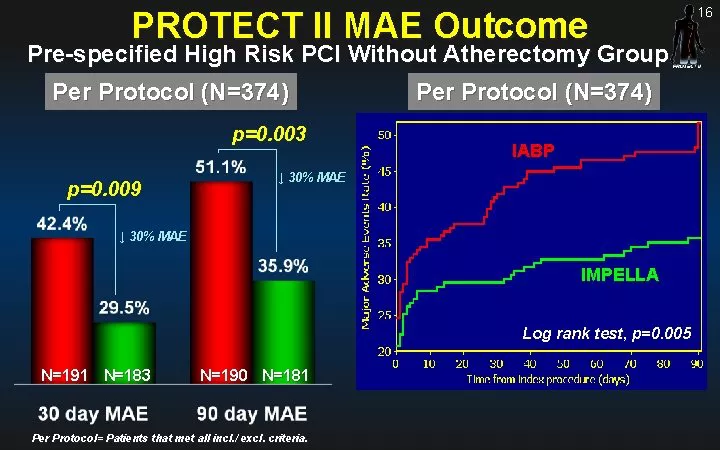
(图:PROTECT II试验结果)
同年,Impella CP® 心脏泵获得美国 FDA 510(k) 许可。Impella CP 是一种微轴泵左心室辅助装置,获准在高风险PCI治疗期间使用数小时,在心源性休克患者中使用长达四天。它2020年获得FDA紧急批准,可用于具有ECMO支持的 COVID-19 患者。
2013年,用于右侧心脏支持的心脏泵——Impella RP在美国开始临床试验。Impella RP能为发生右心衰竭的患者提供临时循环支持。Impella RP在2016年成为唯一获得 FDA 批准的用于右心支持的经皮单通道心脏泵,适用于左室辅助装置植入、心梗、心脏移植或心脏直视手术后出现急性右心衰竭或代偿失调的患者。
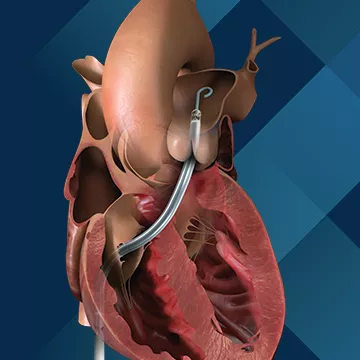
(图:Impella RP)
2014年,接受Impella治疗的患者超过 20,000 名。Abiomed 宣布收购位于德国柏林的医疗器械公司ECP,以扩大和加强现有知识产权和产品平台。ECP当时正在开发一种经皮可扩张导管泵,可通过外部驱动轴增加心脏的血液循环。ECP 拥有 40 多个专利及专利申请,这些专利将添加到 Impella®设备的大约100项专利及专利申请中。
2015年,Impella 2.5® 获得FDA用于选择性和紧急高风险手术的上市前批准(PMA)。2016年,Impella 左侧心脏泵的整个系列获得了FDA用于心源性休克的PMA 批准。Impella CP® 获得FDA用于高风险 PCI 手术的PMA 批准。同年,Impella 2.5® 和Impella 5.0®心脏泵获得日本监管部门批准。
2017年,接受Impella治疗的患者超过 50,000 名。日本医生使用Impella治疗了首例患者。同年,Abiomed位于马萨诸塞州丹佛斯的新扩建总部盛大开业。

(图:公司总部)
2018年,接受 Impella治疗的患者超过 100,000 名。德国医生使用带有SmartAssist技术的Impella 5.5心脏泵治疗了首位患者。SmartAssist®技术通过实时智能改善结果,它应用了光学传感器,无需图像引导即可重新定位泵,且能早期识别右心衰竭。
同年,研究人员宣布STEMI DTU试点随机对照试验结果成功。STEMI DTU 试验是一项前瞻性、多中心、双臂随机对照试验,试验计划招募 668 名接受 STEMI(ST 段抬高型心肌梗死)心脏病发作治疗的患者。临床前试验表明,Impella 心脏泵在冠状动脉再灌注前使用,可减少梗塞面积。
2018年,Abiomed还发起一项女性心脏康复倡议活动,该倡议与公司获得的一项FDA批准相结合,该批复允许Impella用于与心肌病相关的心源性休克,包括女性围产期和产后心肌病 (PPCM))。

2019年,首批美国患者使用 Impella 5.5® with SmartAssist® 进行治疗。该心脏泵专为长期支持而设计,峰值流量高达每分钟6 L,已获得最长 14 天的左心室支持许可,通过使用实时智能使患者活动起来从而优化心脏恢复。
2020年,全球接受 Impella® 治疗的患者超过 150,000 名。FDA 授予Impella为某些 COVID-19患者提供治疗的应急使用授权(EUA)。此外,Abiomed 还收购了 Breethe™ 及其 OXY-1 系统,并用ECMO 技术治疗了首批患者。OXY-1系统,是一项先进的心肺支持系统,带有集成的制氧机,提供独立的氧气源,帮助患者在治疗期间四处走动。新颖的设计,可让医务人员直观进行设置、管理和监控。


(图:OXY-1系统)
2020年,Abiomed还获得一项FDA研究器械豁免(IDE)申请,允许使用9 Fr Impella ECP™ 治疗首个患者。Impella ECP是目前世界上最小的心脏泵,峰值流量大于每分钟3.5 L,通过一个细长护套输送。ECP 代表可扩展心脏功率,目前介入心脏病学家常用的护套或插管, 13 Fr到24 Fr大小不等。Impella ECP使用的护套和泵为9 Fr,在降主动脉中卸下护套,心脏泵可扩展到约18 Fr。专门设计的尾纤,无需导线即可穿过主动脉瓣,并从心室内部泵血。手术完成后,心脏泵缩小并重新套回至9 Fr护套。
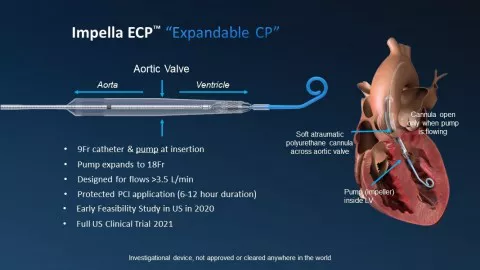
(图:Impella ECP™)
三、公司主要产品及服务
目前,Abiomed公司的产品和技术包括Impella系列心脏泵及Abiomed Breethe OXY-1 System™心肺支持系统。
其中,Impella心脏泵为公司最主要产品,96%收入来源于Impella系列产品。该系列包括上述提到的:Impella 2.5®,Impella CP® with SmartAssist®,,Impella 5.0® & Impella LD® ,Impella 5.5® with SmartAssist®,,Impella RP®。
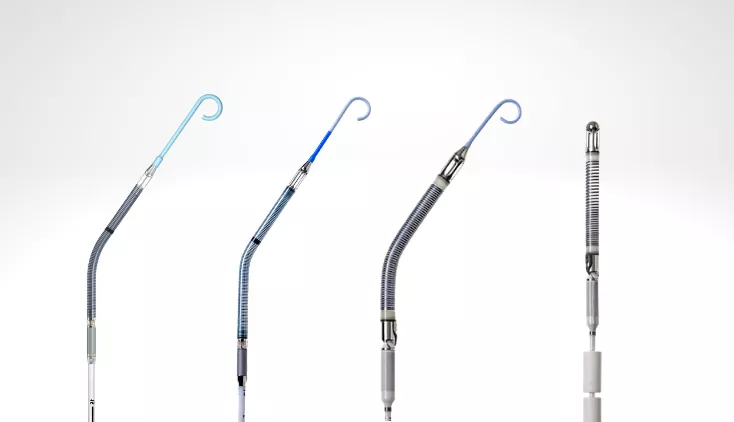
(图:Impella®系列心脏泵)
除产品外,在过去几年中,Abiomed还开展了光学传感器技术、智能设备互联技术(Impella Connect)等新技术的服务。其中,Impella Connect服务,是一项安全、基于云的远程监控技术,能帮助医务人员获得更好的患者结果。
(图:Impella Connect)
根据2021年公司第四季度财报(Q 4 FY 2021),Impella CP®的销售收入占比为77%,是Impella系列中销售收入占比最高的产品,其次为Impella 5.0® /Impella 5.5®,销售收入占比为15%,最后是Impella 2.5®和Impella RP®,销售收入占比分别为4%。
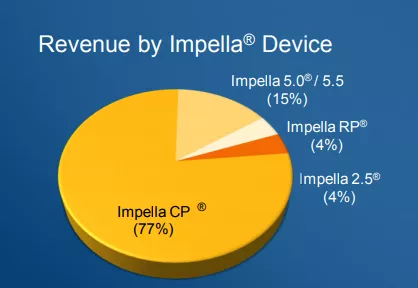
(图:Impella系列销售收入占比)
截止2021财年,上述产品在美国医院的渗透方面,引入Impella CP® 及Impella 2.5®的美国医院达1509家,占比最高。引入Impella 5.0®和Impella 5.5®的医院达663家,引入Impella RP®的医院达588家.
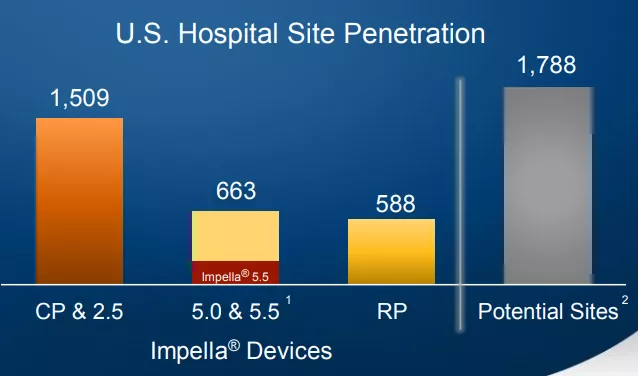
(图:Impella系列美国医院渗透情况)
四、公司全球业务分布及中国业务
目前,Abiomed在全球拥有约1100多名员工,在美国丹佛斯和马萨诸塞州、德国亚琛和柏林、日本东京设有工厂。Impella系列产品在美国、欧洲、日本、加拿大、中国、印度等国家获得了30多项全球监管机构的批准,产品在上述市场的渗透率不断提高,业务版图不断扩大。
根据Q 4 FY 2021财报,公司在美国、德国、欧洲和日本等4个全球主要市场表现良好,日本市场患者数量同比增长50%,销售收入同比增长38%。欧洲和德国市场的销售收入同比增长分别达到28%和31%。
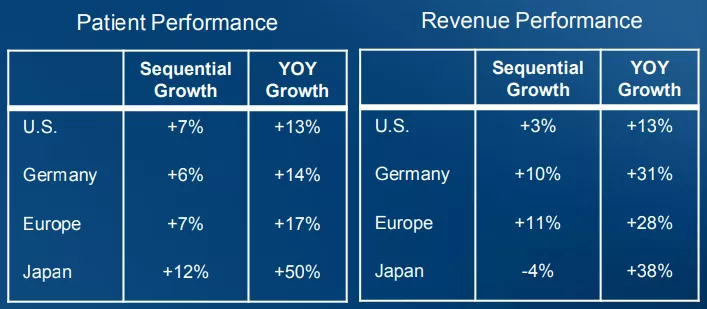
(图:2021财年公司在全球主要市场的增长表现)
2021财年,公司的全球销售收入也呈稳定增长之势,总销售额达8.48亿美元。
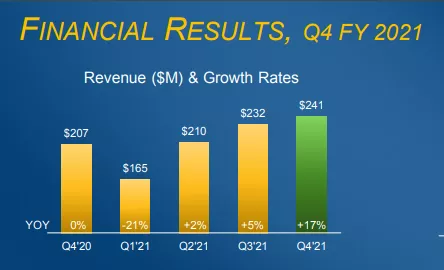
(图:公司FY 2021营业收入)
在中国,Abiomed主要通过代理公司——北京诚诺美迪科技有限公司销售产品。北京诚诺美迪科技有限公司成立于1999年,是以从事高科技医疗器械引进学术交流及培训为主的专业公司。
2006年美国Abiomed与北京诚诺美迪科技有限公司达成合作,不断向国内介绍和推介心衰治疗的理念、技术和产品,特别是近期在国内开展的Impella心室辅助系统的应用,为高危冠脉介入治疗的病人和高危心外科进行手术的病人提供了一种保驾的装置,得到国内医生和患者的好评。
结语
从世界上第一个人工心脏到世界上最小的心脏泵,Abiomed公司成立40年来,向市场推出了一系列领先市场的心室辅助装置(VAD)。截至 2020 年,Impella心脏泵依然是 FDA 历史上研究最多的机械循环支持设备,拥有超过 14 年的研究、超过 140,000 名患者的临床数据以及超过 650 篇同行评审的出版物。不论器械市场如何喧嚣,Abiomed始终致力于为严重心脏病患者开发和提供更小、更智能、更互联的Impella产品,也因此成为了过去十年全球增长最快的企业之一。
来源:思宇医械观察



