




要么第一,要么唯一;微创医疗有望成为中国高值耗材王者?
2021-09-16
微创医疗在几乎所有高值医用耗材所有主要细分赛道上都有布局,要么第一,要么唯一(除骨科外,但未来有反转机会)。
虽然微创医疗2020年财报却并不太靓丽,主要受新冠疫情和冠脉支架受集采政策影响,但市场对微创医疗依然保持乐观态度。
收入结构多元化,抗风险能力进一步提升
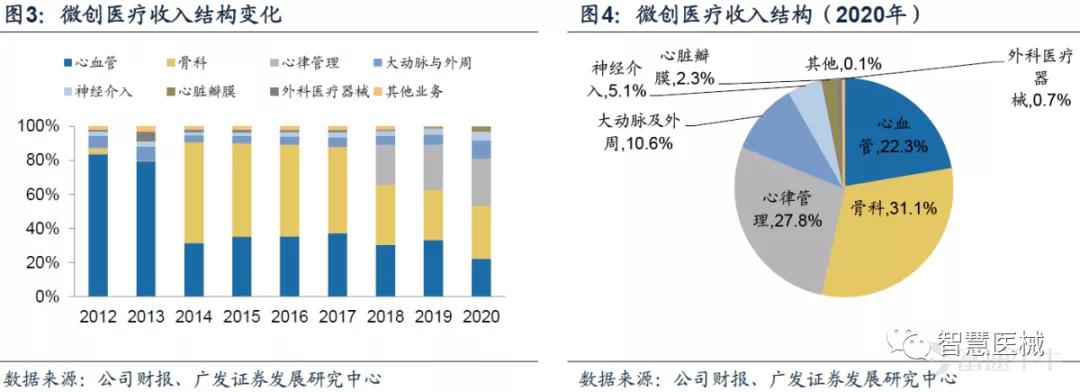
从收入结构来看,微创医疗已经由过去的单一业务发展成为多业务齐头并进的公司,其抗风险能力也进一步得到加强。
其中,心血管业务受疫情影响和集采影响较大占比下降明显,骨科与心律管理业务占比提升。心脏瓣膜业务收入增长383.4%,大动脉及外周血管介入产品业务收入增长40.9%,神经介入产品收入增长17。
1)心血管介入:集采推动放量,新产品和海外市场开拓可期
微创医疗起家于冠脉介入,国内市场份额多年排名第一。冠脉支架类似一个一个管状弹簧网,把狭窄的血管撑开,让管道再通,让心脏满血复活。
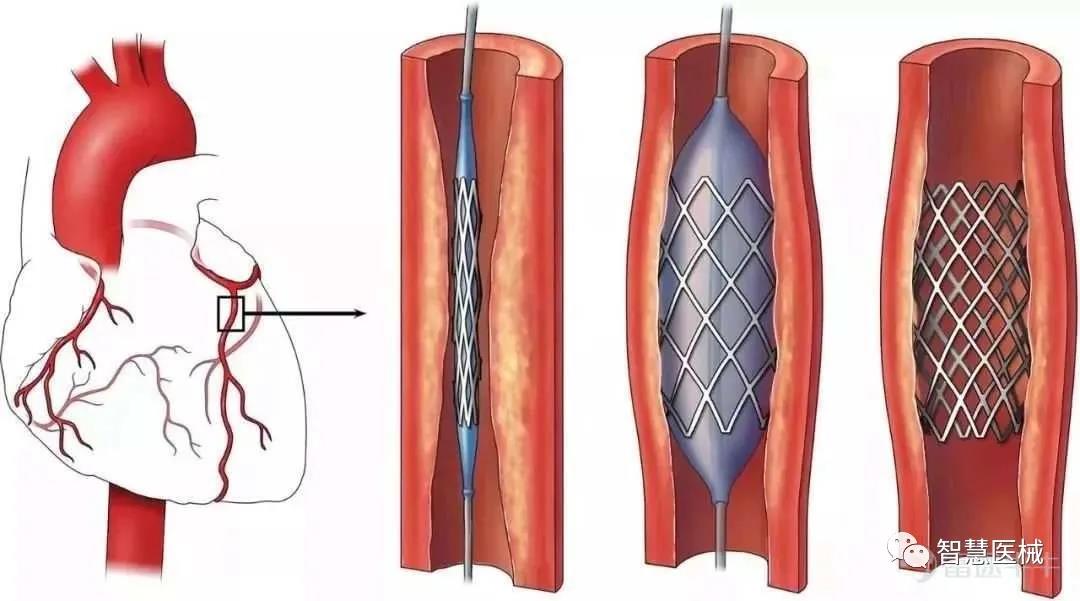
图片来源:微创医疗官网,富途证券整理
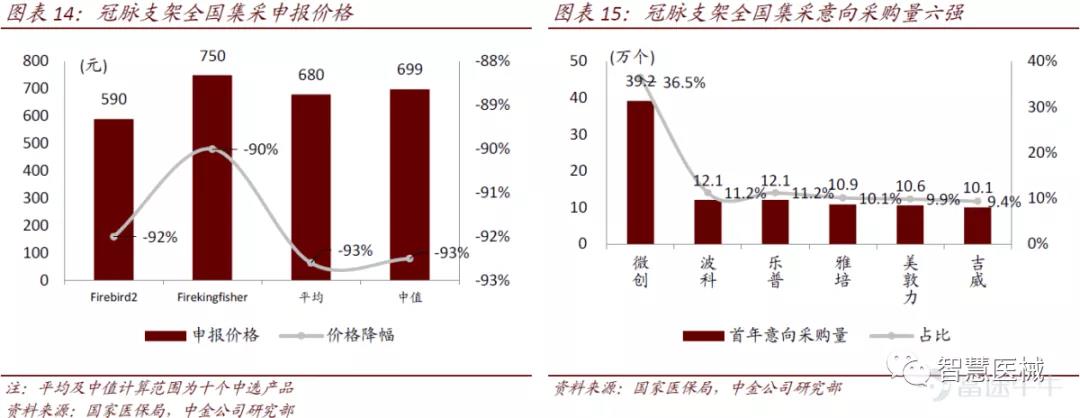
高值耗材集采时代开启后,龙头平台型企业风险抵御能力更优秀。随着国产冠脉支架技术和医生经验的成熟后,2020年冠脉支架成为国内首个高值耗材集采品种,微创医疗两款低端产品Firebird2 和Firekingfisher入围,降价幅度低于平均水平和中值,同时政府采购量最大。
另外,微创医疗高端产品Firehawk 和Firecondor两款靶向药物洗脱支架,直接面向高端自费市场。
高端产品Firehwak冠脉定位不仅仅是中国市场,还积极布局海外市场。2018年9月Firehwak在欧洲进行大规模临床试验,仅用同类1/3的药剂量便同时解决了「低晚期血栓率」和「低血管再狭窄率」10多年来心血管界的这一大难题;
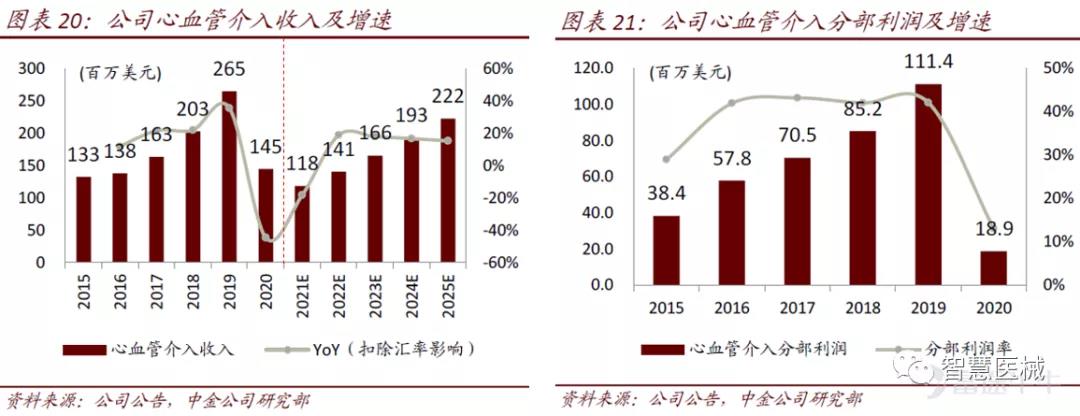
2020年公司心血管介入业务实现销售额1.45亿美元,同比下降44.6%,此次收入下降主要受两大因素影响:1)2020年上半年受疫情影响,手术未能正常展开;2)中国冠脉支架带量采购政策影响,更多患者将选择在2021年手术;
2)骨科:产品组合不断丰富,国产关节增长靓丽
2020年公司骨科业务实现收入2.02亿美元,同比下降13.7%,主要受疫情影响。海外骨科业务收入为1.72亿美元,yoy-16.8%,在Q3季度业绩有明显回升。
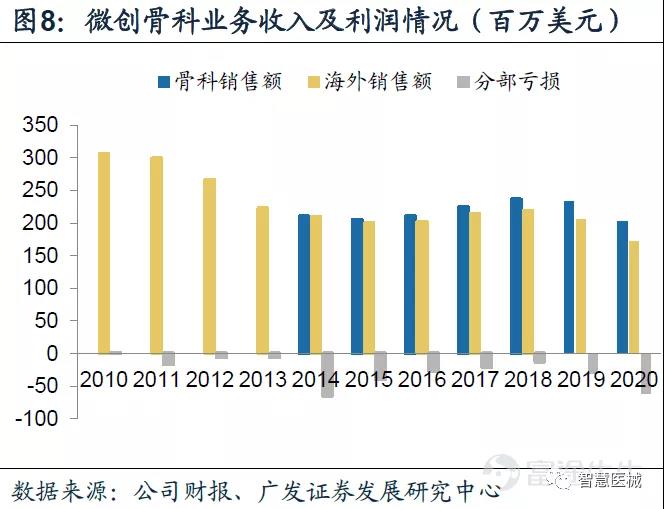
微创医疗骨科业务收入主要来自海外地区,中国业务只有15%。2021年Q2中国将开始第二批高值耗材中骨科集采,微创医疗骨科业务收入主要来自海外,受国内集采政策影响程度小。
MicroPort Orthopedics 已成为全球第五大骨科人工关节生产商,关节领域的产品布局完整。目前有众多关节类产品尚未在中国获批上市,未来领先产品将陆续进入国内市场。
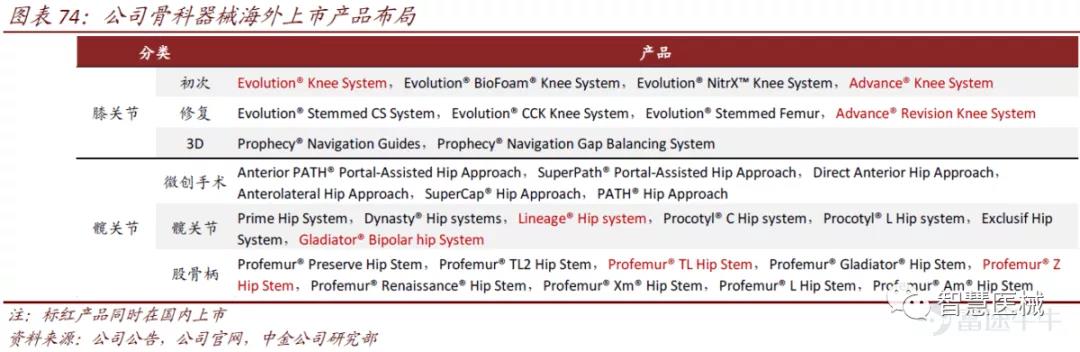
MicroPort Orthopedics 已成为全球第五大骨科人工关节生产商,关节领域的产品布局完整。目前有众多关节类产品尚未在中国获批上市,未来领先产品将陆续进入国内市场。
图片来源:潍坊市市立医院
2020年公司心律管理业务实现收入1.80亿美元,较2019年同期下降16.2%,主要是受海外疫情的影响。从下图得出,2020年国际业务收入0.92亿美元,yoy-5.7%,而中国业务同比增长15.5%。
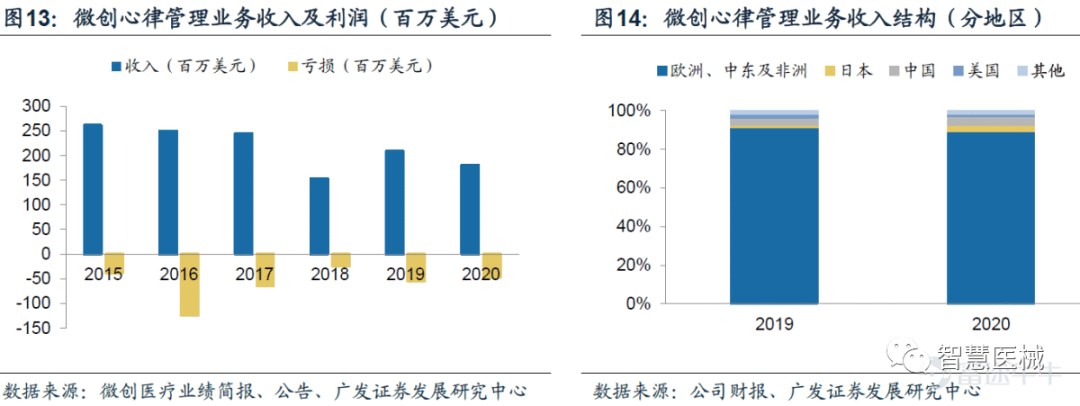
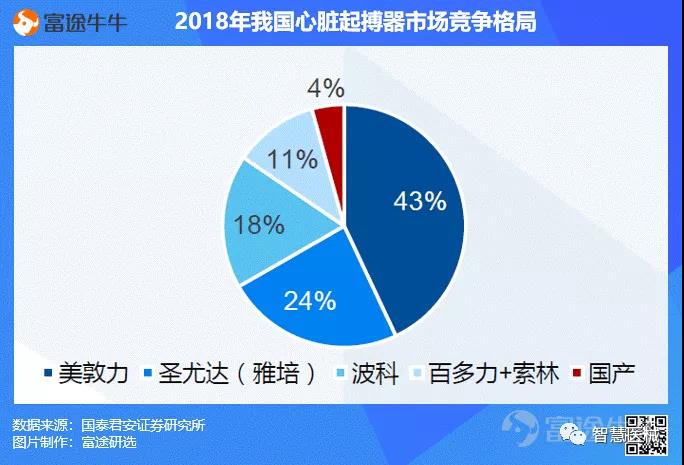
我国心脏起搏器渗透率低,国产产品市场份额不足5%,潜在空间广阔。目前国内起搏器市场规模45亿,由于研发生产制造壁垒高,进口品牌占了95%的市场份额。国产仅有三家企业:乐普(秦明)、微创(创领,与索林合作)和先健(与美敦力合作)。
微创心律在国产心脏起搏器品牌份额稳占第一,截至2020年底,已进入480家医院,新增168家医院。
3)大动脉及外周:主动脉行业领军者,2021Q1季度再提速
国内主动脉患者近200万,2022年介入治疗市场规模可达20亿。目前国内主动脉介入手术约为3万例左右,未来随着国产支架的普及,未来还有非常大的增长空间。
图片来源:医药卫生报,富途证券整理
2020年公司大动脉及外周血管介入产品收入6849万美元,同比增长40.9%。根据心脉医疗财报,心脉2021Q1实现收入1.97亿人民币元,同比增长高达99.1%。
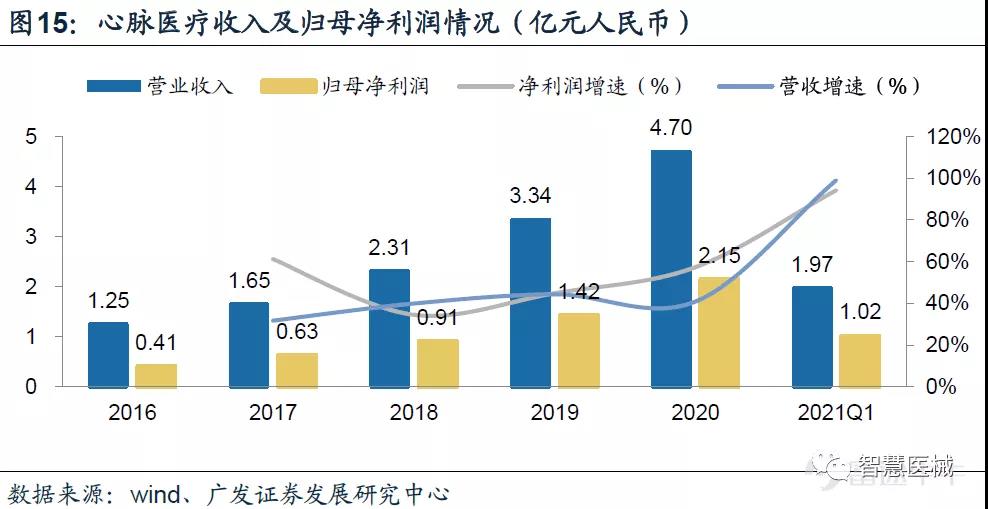
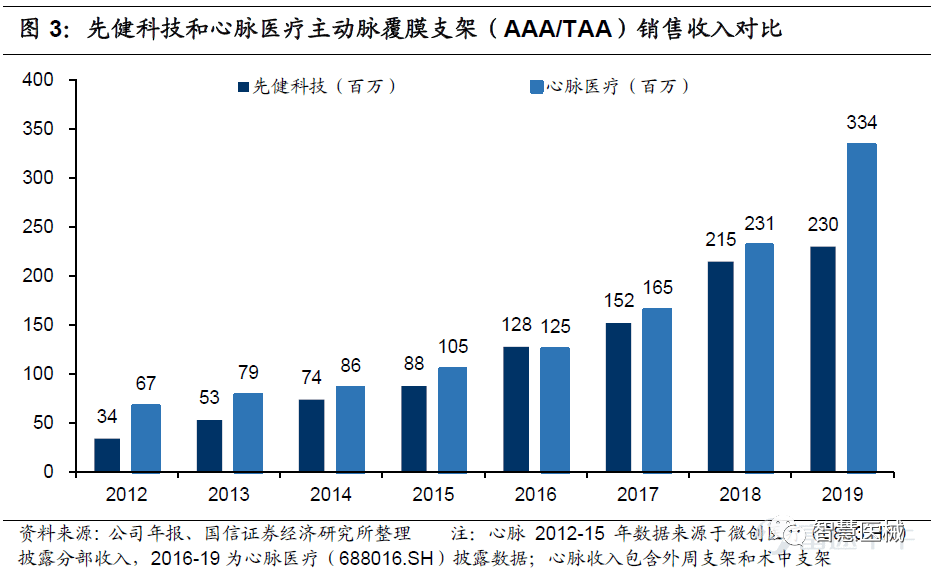
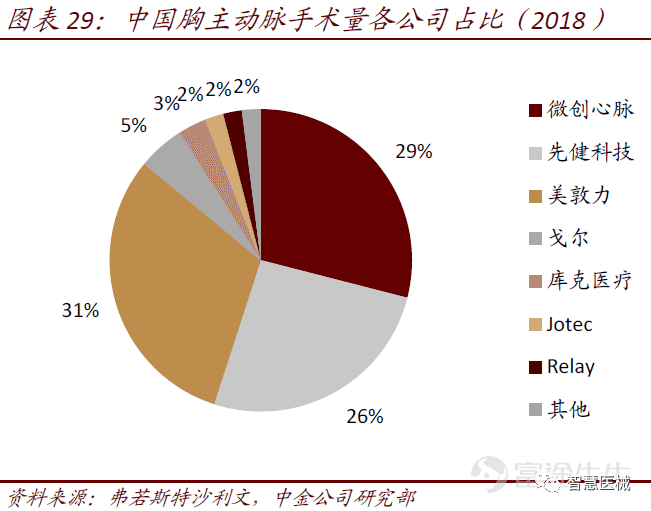
微创心脉在国内最大竞争对手是先健科技。全球主动脉支架市场三巨头分别是美敦力、库克和戈尔,国产厂商的领导者主要为先健科技和微创心脉。微创心脉产品迭代快、产品组合均强于先健。
值得一提的是,Fontus 分支手术支架植入系统和Talos 胸腔支架植入系统有望今年获批,未来有望进一步抢占国内的市场份额。
4)心脏瓣膜:TAVR 销售强劲,研发稳步推进
TAVR介入手术属于微创手术,创伤伤口小,恢复时间短,手术风险低,尤其适合高龄75岁以上高龄老人。TAVR手术时间长为1-2小时,而开胸手术时常为3-6小时。
数据来源:启明医疗招股说明书,富途证券整理
VitaFlow系列产品未来市场前景广阔,国内仍处较早期阶段。目前公司核心产品是VitaFlowTM 和 VitaFlow II产品,均属于TAVR领域。VitaFlowTM 已于2019年7月获批上市,二代产品VitaFlow II 预计2021年底前在中国获批上市。

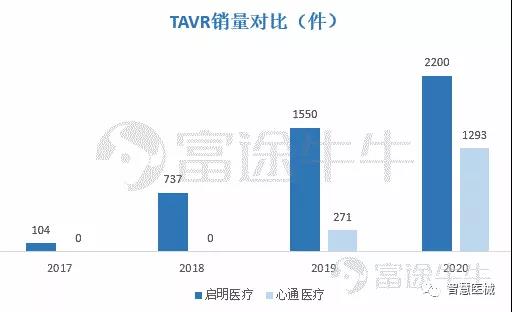
TAVR手术在国内远未满足,预计2021年心通医疗VitaFlowTM销量超越启明医疗。目前我国TAVR领域呈现「3国产+1进口」局面,但心通的对手只有启明医疗,且心通TAVI产品后续爆发力可能更强。
虽然心通医疗产品在2019年才上市,但背靠国内微创手术领域龙头(微创医疗)渠道优势,VitaFlowTM销量快速进入国内医院。
TAVR手术在国内远未满足,预计2021年心通医疗VitaFlowTM销量超越启明医疗。目前我国TAVR领域呈现「3国产+1进口」局面,但心通的对手只有启明医疗,且心通TAVI产品后续爆发力可能更强。
虽然心通医疗产品在2019年才上市,但背靠国内微创手术领域龙头(微创医疗)渠道优势,VitaFlowTM销量快速进入国内医院。
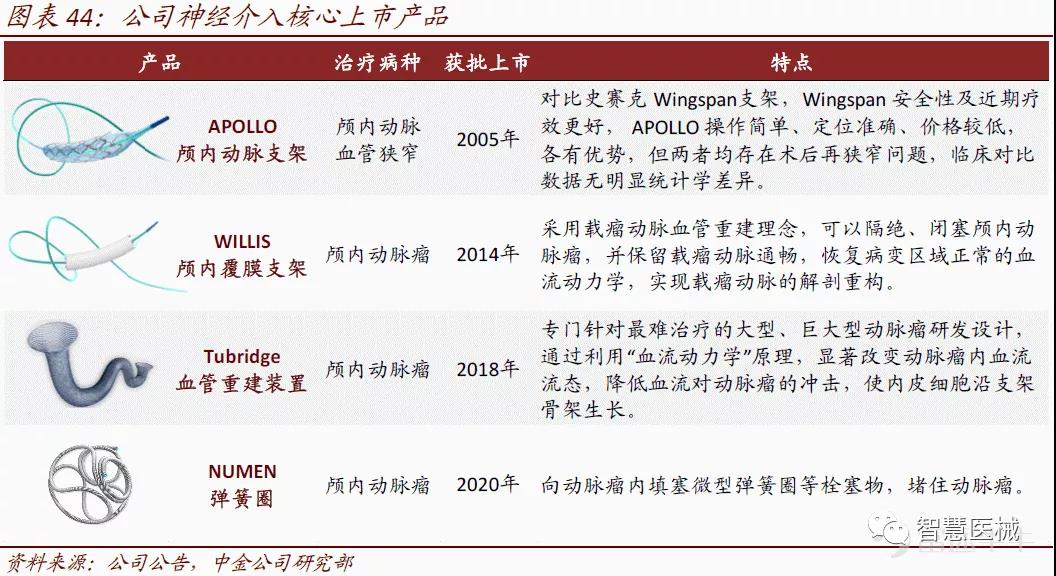
5)神经介入:布局完整产品线,Tubridge打破了进口密网支架垄断地位
神经介入一直被誉为介入手术皇冠上的明珠。神经介入属于医学非常重要的部分,难度极大、范围广,被誉为介入手术皇冠上的明珠。
我国目前神经介入市场80%以上是被外资垄断着,未来国产替代机会非常大。美敦力是全球神经介入的绝对类龙头,国内60%以上的市场被美敦力占据着,而史塞克、Penumbra其他在细分赛道中各占据领先地位。

微创医疗主要布局神经介入完整的产品线,是国内绝对的龙头。APOLLOTM支架自上市以来凭借其出色的安全性和有效性,不断夯实在缺血性中风治疗领域的绝对领导地位。Tubridge是为数不多有能力与外资产品竞争相关细分市场份额的国产神经介入产品,它打破了进口密网支架的市场垄断地位,与美敦力的 pipeline 密网支架并称为国内密网支架市场的「双雄」。
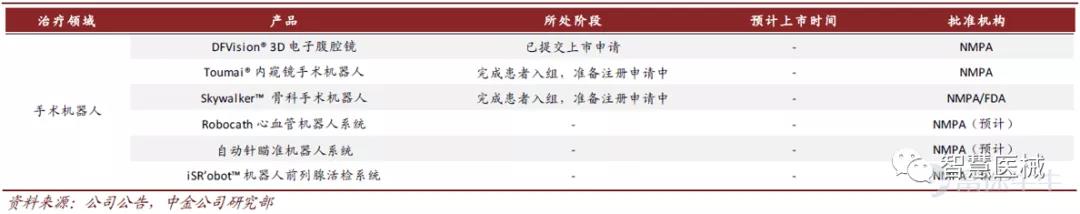
6)手术机器人:多赛道布局,即将进入商业化收获期
手术机器人研发稳步推进,即将进入商业化收获期,同时也填补了我国手术机器人的空白。公司自主研发的蜻蜓眼DFVision三维电子腹腔镜成功豁免临床,并已于报告期内递交注册申请,预计今年获批上市。
另外,图迈Toumai手术机器人于2021年1月已完成患者入组,成为了首个在泌尿外科领域完成多领域的国产腔镜手术机器人。鸿鹄Skywalker骨科手术导航定位系统于2020年进入绿色通,也在报告期内完成了患者入组工作。图迈Toumai手术机器人和鸿鹄Skywalker手术机器人预计在2022-2023年获批上市。
中金给予微创医疗目标价为70港元
考虑到公司的产品仍处于亏损状态,同时潜在上市产品未来有望提供稳定的现金流,中金证券采用DCF估值,给予微创医疗的目标价约为70港元。

小结
微创医疗几乎在所有前沿的高值医用耗材的细分赛道上做到国产第一,如微创心脉反超并甩开了先健,成为第一,心通将反超启明成为第一,微创机器人国产第一,神经介入(神通)国产第一(国内所有对手营收都比神通小一个量级),心律国产第一。中金近期给予微创医疗目标价约为70港元。
来源:智慧医械



