




Syncardia人工心脏,为晚期心力衰竭患者架起生命桥
2021-11-12
对于终末期心力衰竭患者而言,心脏移植是最佳的治疗手段,但人体心脏供不应求。
据《中国心血管健康与疾病报告2019》,保守估计我国约有400万慢性心力衰竭患者。而全国35家(不含港澳台)心脏移植中心,2015年到2018年共完成心脏移植1583例,接受心脏移植的人数要远远少于晚期心力衰竭患者的人数。在等待心脏移植的过程中,一些患者很有可能会发病死亡。
这种情况下,患者可以考虑暂时植入人工心脏以维持生命,等待合适的心脏供体。Syncardia研发的人工心脏是世界上唯一获FDA批准上市的全人工心脏,它被用作晚期心力衰竭患者心脏移植前的过渡治疗,在临床上已使用超过35年。
作为过渡性治疗手段,Syncardia的人工心脏为晚期心力衰竭患者架起了一座生命桥。
首个获FDA批准的全人工心脏
心脏最核心的功能就是泵血,而心力衰竭患者由于心肌舒张和(或)收缩功能障碍,不能实现正常的心脏泵血。人工心脏就是用生物机械手段部分或完全替代心脏的泵血功能,来维持患者全身的血液循环。
人工心脏按功能可以分为心室辅助装置(VAD)和全人工心脏(TAH)。心室辅助装置是一种用于辅助心脏循环的机电泵,安装在患者心脏周围,而全人工心脏则是用新的心室和瓣膜来替换患者的自然心脏。
Syncardia公司一直致力于研发全人工心脏,其心脏产品起源于历史上第一颗永久性人工心脏Jarvik 7,已经拥有30几年的历史。
1982年,心脏病患者Barney Clark首次植入了Jarvik 7,并且成功地存活了112天。之后在1985年到1991年期间,大约有170例患者使用Jarvik 7作为他们心脏移植前的过渡治疗。但由于Jarvik 7的两个心室通过数根电线连接到体外,所以患者在植入后容易发生败血症和多器官功能衰竭。1991年,FDA叫停了这款人工心脏的临床应用。
此后,Syncardia公司的前身——CardioWest公司对这款人工心脏进行了改良,并重新命名为CardioWest。2004年,CardioWest人工心脏获FDA批准作为不可逆的双心室衰竭患者心脏移植前的辅助治疗手段,成为了当时唯一一款获FDA批准和CE认证的全人工心脏。
17年过去,Syncardia的全人工心脏仍然是全球唯一获FDA批准上市的产品。为了更好地研发、生产和销售全人工心脏产品,公司正在不断地调整人才队伍。
Syncardia最初成立于2001年,初创成员有Jack G.Copeland、生物医学工程师Richard G.Smith、介入心脏病专家Marvin J.Slepian。2016年9月,该公司被私人股权投资公司Versa资本管理有限公司收购。
Don Webber于2018年加入Syncardia,负责制造运营、工程、质量和供应链物流的首席运营官。因为在工作中的出色表现,他于2019年晋升为首席执行官。Don在生命科学领域拥有超过25年的管理经验。此前,他曾担任医疗器械公司OptiScan的首席运营官和巴德公司(C.R.Bard,被BD收购)的制造运营副总裁。同年,Blago Herrera被任命为Syncardia的质量副总裁,他曾在OptiScan公司担任高级运营总监。
此外,Syncardia拥有出色的销售队伍。Eric Lambert是公司的欧洲销售主管,他拥有超过18年的医疗保健行业全球销售和营销经验。此前,他曾担任Sorin集团销售和营销主管。John Arancio是公司的北美销售主管,他在医疗保健行业拥有30几年的销售经验。加入Syncardia之前,他在圣犹达医疗担任过各种销售团队的领导。
两款全人工心脏、两款体外驱动器,适应更多患者和场景
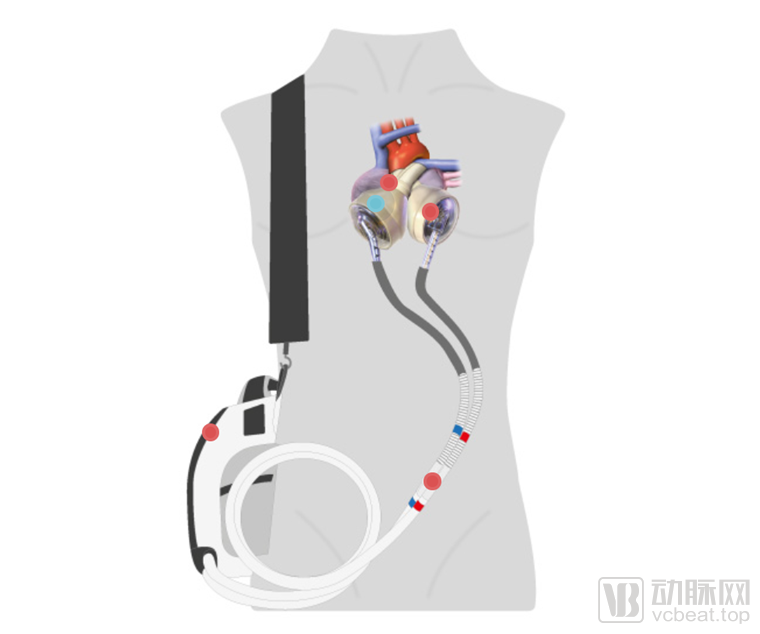
来源:Syncardia官网
Syncardia的全人工心脏由两个心室、四个瓣膜组成。其中两个心室通过尼龙拉带连接在一起,外科医生可以根据患者的解剖结构将心室放置在胸部,而四个瓣膜则控制着血流进入和流出心室的方向。它是由一种特殊的生物相容性塑料制成,具有高度的抗疲劳性和强度,可以避免患者对植入物产生排异反应。
这种全人工心脏运作的原理是气动驱动。体外的气动泵能够产生空气和真空脉冲,对每个心室内的隔膜进行充气和放气,以推动血液进出心室。
此外,SynCardia 的人工心脏是一个固定速率的设备,在设置好节拍后,每分钟的节拍都将保持不变。当患者处于运动状态时,他的身体会自动将更多的血液流到人工心脏,而人工心脏会相应地将增加的血液回流到身体。
目前Syncardia公司共有两款人工心脏、两款体外驱动器,适合更多的晚期心力衰竭患者和治疗场景。
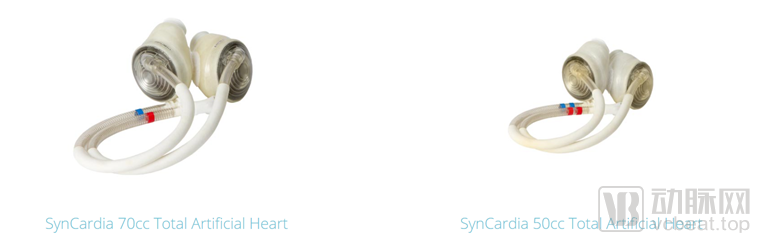
来源:Syncardia官网
SynCardia 70cc是世界上首个上市的全人工心脏,被FDA批准用于终末期心力衰竭患者,作为他们心脏移植前的过渡手段。它在全球拥有1700多个移植患者,他们的年龄上至80岁,下至9岁。70cc TAH体积较大,适合大多数的男性和一些女性。它每分钟能够产生高达9.5升的血流,而通常情况下一个普通人在安静时每分钟的泵血量约为5升。
目前,70cc TAH正在进行FDA批准的调查设备豁免(IDE)临床试验,针对没有资格接受心脏移植的成年患者进行治疗。这或许是Syncardia向着永久性人工心脏迈出的关键一步。
50cc TAH是70cc TAH的较小版本,为大多数的妇女和一些青少年设计,它每分钟能够产生7.5升的血流量。这款产品使得更多的妇女和青少年能够受益于全人工心脏这一技术,扩大了移植患者的范围。
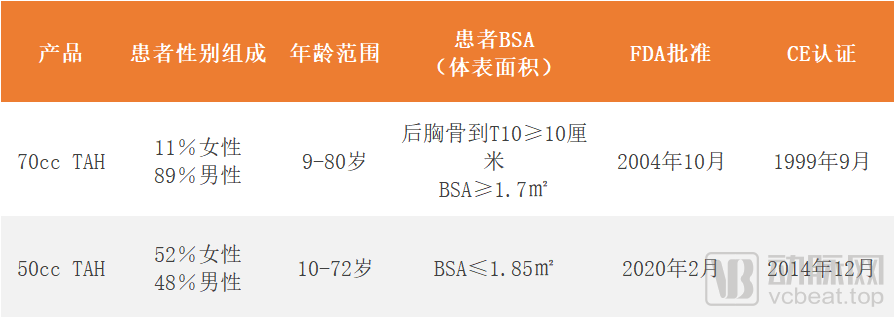
根据SynCardia公司对患者的人口统计学调查,50cc TAH的移植患者中,女性所占比例相较于70cc TAH而言有明显的提升,同时18岁以下的儿童患者数量也有提升,占到了总移植人数的22%。

来源:Syncardia官网
Syncardia的人工心脏通过两根穿过腹壁的插管与体外的驱动器相连。为了方便患者的术后行动,Syncardia根据不同的应用场景研发了两款驱动器。
其中,C2驱动器供患者在医院疗养时使用。医务人员可通过C2驱动器向两个心室提供独立、可调节的真空和压力,根据每个患者的不同需求来定制驱动器的性能。在使用过程中,C2驱动器可以放置在医院的推车或盒子里,以便于运输和充电。
在患者术后恢复的初期,C2驱动器可放在医院推车内。推车配有大型触摸屏显示器、支撑把手和锁定脚轮,能够在患者行走时提供牢靠的支撑。而在患者可以下床行动后,C2驱动器又可放在相对较小的盒子中,方便病人行走、锻炼。
当患者病情稳定可以出院时,SynCardia还提供一款自由便携式驱动器。这是一种更小、更轻的气动泵,重为13.5磅(约6.1公斤),可放在患者背包里随身携带。等待心脏移植的过程十分漫长,患者能够带着人工心脏在院外自由活动,不仅是提高了他们的生活质量,还可以省下一笔住院费用。
离开固定的医疗环境,适应多变的生活场景,这是人工心脏模拟人体自然心脏的重要一步。
国产心室辅助装置很有可能赢取本土市场
高难度的技术、较难展开的临床试验使得全人工心脏的发展始终保持低速度。
2020年末,法国Carmat公司获欧盟CE认证,其全人工心脏产品能够作为晚期心力衰竭患者心脏移植前的过渡工具。经过十几年的研发试验,该公司的Aeson人工心脏将于今年第二季度上市。
尽管全人工心脏已经投入商用,技术上也在逐渐进步,但全球的总体移植数还是很少。首要原因就在于高昂的价格。人工心脏加上手术、术后排异药物的费用,这是一笔不小的开支。其次是心脏移植的不可逆性。安装全人工心脏之前会摘除掉患者原有的心脏,这个过程是不可逆的,很多人在心理上还是不敢用一颗机械心。
再就是生活上的不便,全人工心脏必须时刻有电,一旦电量耗尽,患者就会濒临死亡。而且患者需要随身携带外部的驱动器,有许多生活上的禁忌,不能碰水、剧烈运动等。
正是由于全人工心脏面临技术壁垒、伦理挑战等问题,目前在人工心脏领域中应用得更多的是心室辅助装置。与全人工心脏不同,植入心室辅助装置无需摘除人体原有的心脏,在心理层面上更容易被大众接受。
目前,中国企业正在积极研发心室辅助装置。国内第一款也是唯一一款上市的心室辅助装置是重庆“永仁心”人工心脏。它是重庆永仁心医疗器械有限公司引进日本HI-LEX集团的技术来生产的植入式左心室辅助系统,填补了国内人工心脏领域的空白。
而苏州同心医疗器械有限公司自主研发出拥有完全自主产权的全磁悬浮人工心脏,重量不到180克,大小和乒乓球差不多。除了在确保运行效率的同时尽量缩小人工心脏中血泵的体积,同心医疗还解决了血液相容性的问题,有效避免了患者出现血栓。
目前为止,NMPA仅批准了2例关于人工心脏治疗终末期心脏衰竭安全性和有效性评价的注册登记临床试验研究,分别是永仁心和同心的人工心脏。这两家公司走在了中国心室辅助装置研发的前列。
尽管美国有雅培、美敦力等产业巨头,已经形成了较为成熟的心室辅助装置市场。但目前这些高价产品还没有进入中国,国内这个领域尚存在一片蓝海。国产人工心脏有很大机会赢取本土市场。
来源:动脉网



