




最新!医疗器械生物学评价对医用材料的要求
2018-11-15
11月13日,国家食品药品监督管理总局医疗技术评审中心,根据原国家食品药品监督管理总局2018年度医疗器械注册技术审查指导原则项目计划的有关要求,组织编制了《医疗器械生物学评价指导原则第1部分:总则》,现已形成征求意见稿,即日起在网上公开征求意见。

本指导原则的制定,旨在指导医疗器械行业对与人体直接或间接接触的医疗器械进行合理充分的生物学评价,以确定人体与器械部件材料接触是否会产生毒性。
在对新器械进行评价时,如果器械不与组织直接或间接接触,申请人应明确声明,且无需提供进一步的生物相容性信息。
对与人体直接或间接接触的医疗器械进行生物学评价,应遵循以下基本要求:
器械制造材料的选择及其生物相容性评价,应首先考虑直接或间接组织接触的可能性以及任何关于器械制造的有用信息(例如,每项组件材料的化学配方,包括粘合剂、已知和疑似杂质、以及与加工相关的成分)。对于由纳米材料组成、包含纳米材料或会生成纳米材料的医疗器械,由于纳米材料潜在的特有性质,可能需要对其生物学评价进行特殊考虑。
另外,直接或间接接触医疗器械的包装材料可能将化学物转移到医疗器械上,然后间接转移给患者或者临床医生。因此,医疗器械的生物学评价还应关注产品的内包装材料可能引起的风险。
应考虑制造材料、成品、可能沥滤的化学物质或降解产物与器械总体毒理学评价的相关性。如有证据显示,特定的物理特性对生物相容性有影响,还应关注产品的物理特性。应关注的物理特性包括但不限于孔隙率、粒径、形状和表面形态。
与生物相容性评价相关的终点应考虑医疗器械与人体接触的性质、程度、频次、时间和条件。可利用该原则对器械进行分类,以便于在总体生物相容性评价中选择合理的终点。
境内申请人提供的医疗器械生物学评价报告中含有的生物学试验报告应由具有相应生物学试验资质的检测机构出具;境外申请人提供的医疗器械生物学评价报告中含有生物学试验报告的,企业应提供生物学试验室所在国的GLP证明。

应提供完整的试验数据,且此数据可用于独立做出结论。
申请人应在医疗器械的整个生命周期内评估其生物安全性。对于可重复使用的产品,应对其最大验证处理周期数的生物安全性进行评估。
如果器械的化学成分、制造工艺、物理结构(如大小、几何构型、表面特性)、预期用途或初级包装发生任何变更,应针对生物相容性是否发生变化以及是否需要进行额外生物相容性试验的情况进行评价。
在评价器械改良情况时,如果改良不会变更任何与组织直接或间接接触的组件,申请人应明确声明,且无需提供进一步的生物相容性信息。但是,如果变更会对其他未经变更的、与组织直接或间接接触的器械部件造成影响,应进行生物相容性评价以确定变更可能造成的影响。例如,如果新增不与组织接触的内部组件,但是,需要通过加热使其与其它患者接触的组件相连,那么在加热时,可能影响与患者接触的组件,从而生物相容性也会受到影响,应进行评价。
在根据本指导原则进行生物学评价时,应联合考虑其他非临床研究、临床研究和上市后经验信息,以整合所有有用的相关信息进行安全性评估。
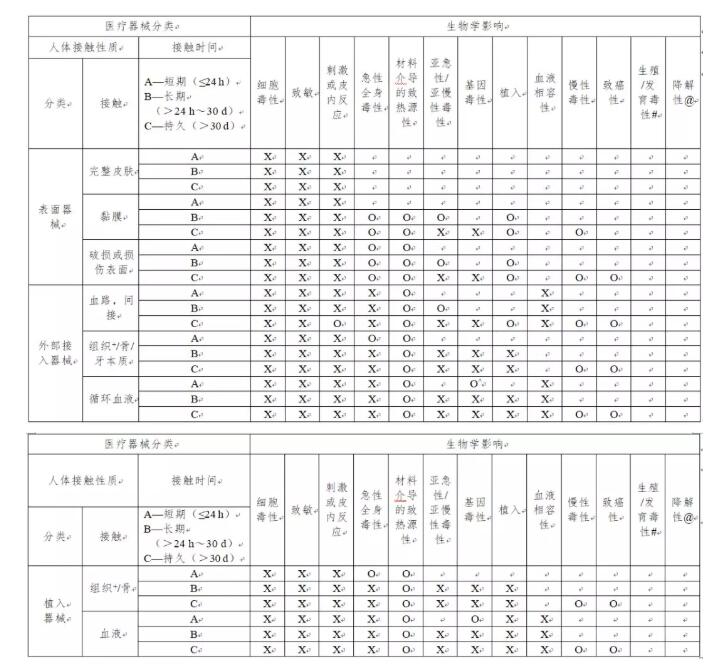
表1 生物相容性评价终点
X代表GB/T16886.1-2011推荐考虑的终点
O代表额外推荐考虑的终点
备注* 应在生物学安全性评价中,利用现有数据、通过额外特定终点试验或针对为何无需对终点进行额外评价的原理来解决所有X和O。
备注+ 的组织包括组织液和皮下间隙。
备注^适用于所有的在体外循环中使用的器械。
备注# 表示对于新材料、已知具有生殖或发育毒性的材料、具有相关目标人群(如孕期妇女)的器械和器械材料在生殖器官中有局部暴露的可能性的器械,应评价生殖和发育毒性。
备注@ 持续与组织接触的预期可降解的任何器械、器械组件或材料应提供降解信息。
如上述表1所述,建议在比GB/T 16886.1-2011概述的范围之外的更大范围中考虑急性全身毒性、亚慢性毒性和植入终点。
建议应考虑在比GB/T 16886.1-2011概述的范围更广的器械/组织接触范围中进行刺激评价。
建议应考虑在比GB/T 16886.1-2011概述的范围更广的器械/组织接触范围中进行遗传毒性评价。
此外,建议申请人考虑通过单独评价来评价可能存在热原的器械材料化学成分。认为这种经材料介导的热原是GB/T 16886.1-2011中急性全身毒性的子集合。但是,如果急性全身毒性或植入研究不包含定期温度测定数据(例如,最初3个小时,每隔30分钟进行一次测定)或未在合理的动物模型中进行该研究(即,家兔),利用该研究数据来替代单独进行的热原评价是不合理的。
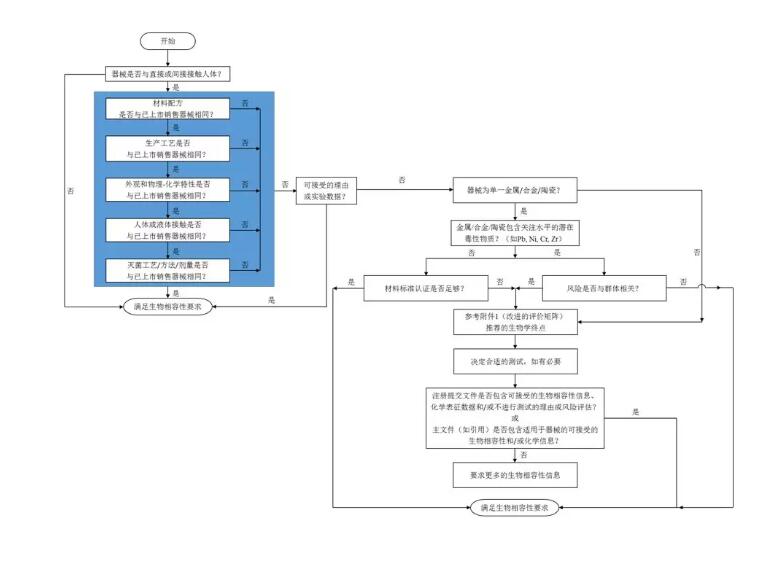
1. 风险评定(Risk assessment):包括风险分析(系统运用可获得信息来识别危害并估计其风险)和风险评价(将估计的风险和给定的风险准则进行比较,以确定风险可接受性的过程)的全过程。
2. 生物相容性(Biocompatibility):某一医疗器械或材料在特定应用中具有适宜宿主反应的能力。
3. 接触(Contact):
a. 直接接触(Direct contact):适用于与人体组织进行物理接触的器械或器械组件的术语。
b. 间接接触(Indirect contact):适用于在液体或气体与人体组织进行物理接触之前所流经的器械或器械组件(在这种情况下,器械或器械组件本身不与人体组织发生物理接触)的术语。
c. 不接触(Non-contact):适用于不直接或间接与人体接触(例如,独立软件或数据库)和适用于除确认不与人体接触外无需生物相容性信息的器械或器械组件的术语。
d. 瞬时接触(Transient contact):适用于与人体组织发生极短暂接触的器械或器械组件的术语(例如,使用不超过一分钟的皮下注射针)。
4. 最终成品(Final finished form):适用于经历所有为“待销售”器械进行的制造工艺的器械或器械组件,包括包装和灭菌(如适用)。
5. 材料(Material):物质或组成或制成物品的物质。
6. 新型材料(Novel material):之前未在任何在中国合法销售的医疗器械中使用的材料。
7. 降解(Degradation):器械分解,可能形成新的化学物质或吸收材料,造成器械(器械功能)的机械特性和/或物理特性随时间丢失。
8. 可浸提物(Extractables):某一医疗器械或材料用浸提溶剂和/或在至少与预期临床使用相同或更严格的条件下浸提时,能释放出的物质。
9. 极限浸提(Exhaustive extraction):随后的浸提至浸提液中的可浸提物质的量小于第一次浸提液中10%检出量的浸提。
10. 可滤沥物(Leachables):医疗器械与水或使用中有关的液体作用时,从该医疗器械释放出的化学物质。
11. 毒理学危害(Toxicological hazard):某一化合物或材料导致不良生物学反应的潜能,考虑该反应的性质和诱发反应所需的剂量。
12. 毒理学风险(Toxicological risk):针对特定的接触水平,发生特定程度不良反应的概率。
13. 中毒(Toxic):造成损伤或死亡(特别是通过化学方法)的后果。
14. 毒性(Toxicity):物质引起中毒的程度。
15. 体内动物研究(In vivo animal study):设计旨在为器械安全性、用于生命系统的潜在性能和/或对器械的生物学反应提供初始信息的非临床动物研究。
16. 申请人(Sponsor):制造商、提交者或申请者。



