






巨头67亿美元入场,达芬奇不再是一枝独秀,手术机器人明年有望进口替代
2019-12-13
1999年,首台达芬奇手术机器人问世,迄今过了20年。
达芬奇手术机器人并非万能,主要应用在心脏、胸、泌尿、妇科、结直肠、儿科和普通外科等学科,但在这20年间的巨大成功使其成为了手术机器人的代名词。
全球66个国家及地区部署了超过5000台达芬奇手术机器人,每30秒就有一位主刀医生利用达芬奇手术机器人开展一例手术。迄今,达芬奇机器人已经在全球完成了600多万例手术。其中,仅2018年就进行了100万例。
研发达芬奇手术机器人的直观手术(Intuitive Surgical)市值高达600亿美元,看上去无法撼动。尽管如此,美敦力、强生、西门子、史赛克等全球医疗器械10强大佬们仍然在2019年前赴后继进入了这个领域。根据统计,四家公司在1年内豪掷67亿美元砸向手术机器人。
那么,究竟手术机器人到底有多火?(微信号:vcbeat)对2019年手术机器人行业进行了盘点。
达芬奇不再一枝独秀
一年前的2018年12月,全球医疗器械的龙头企业美敦力以17亿美元收购Mazor Robotics及其机器人辅助手术平台。这次收购成为了当时医疗机器人领域规模最大的收购案。
一个月后,该公司就在美国推出了Mazor X Stealth脊柱手术机器人,将Mazor Robotics的机器人引导系统技术与美敦力的StealthStation手术导航技术无缝结合。
美敦力此次收购的目标很清晰,即通过此次收购进一步夯实美敦力在全球脊柱和肌肉骨骼治疗领域的强势地位。通过将旗下脊柱植入物、导航和术中成像技术与Mazor Robotics的机器人辅助手术(RAS)系统相结合,美敦力打算为手术的计划、执行和确认,提供一套整合的脊柱手术解决方案。
在收购Mazor Robotics不到一年后,美敦力于2019年9月推出了全新的Hugo RAS手术机器人。这意味着美敦力基本完成了对Mazor Robotics技术的消化,使其手术机器人的商业化进程更进一步。
美敦力虽然涉足机器人较晚,但凭借这次收购后来居上。从宣布收购Mazor Robotics到Hugo RAS的推出,时间跨度不超过1年。美敦力雷厉风行的作风让人印象深刻,这恐怕也是其能够在近年超越器械领域多年霸主强生,成为新的医疗器械龙头的重要原因。
一石激起千层浪,美敦力的这次收购引发了连锁反应。
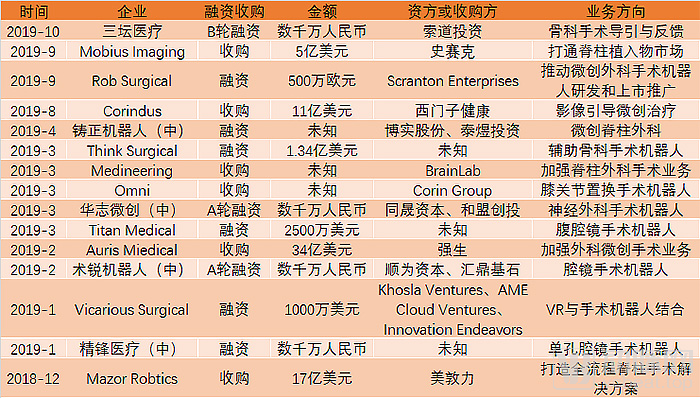
首先应战的是强生。2019年2月,强生子公司Ethicon以34亿美元收购了Auris Health及其获得FDA认证的Monarch手术机器人。这笔交易刷新了美敦力刚刚完成的收购,成为有史以来规模最大的医疗机器人并购交易。
2019年8月,西门子医疗以11亿美元收购手术机器人公司Corindus Vascular Robotics。这家企业主要开发用于介入治疗的CorPath远程手术机器人。这一手术机器人也是首个经FDA批准用于辅助经皮冠状动脉介入治疗(PCI)程序的医疗设备,以帮助医生在放置支架时提高精度和准确度。
仅仅一个月以后,史赛克宣布以5亿美元收购Mobius Imaging及其子公司Cardan Robotics。这次收购使史赛克获得了Mobius的Airo CT移动诊断成像设备,及Cardan Robotics旗下与之配合执行内窥镜脊柱手术的Orian手术机器人,从而可以提供从影像到导航到手术机器人的一站式解决方案。
根据统计,上述四家前十大医疗器械公司在1年内豪掷67亿美金砸向了手术机器人,并且在很短的时间内两度刷新交易纪录。手术机器人在2019年的热度由此可见一斑。
与此同时,2019年还有数家手术机器人初创企业完成了融资。当然,由于较高的技术门槛、较长的研发周期,处于初创阶段的手术机器人企业在融资额上并不那么突出。不过,仍然也有Think Surgical这样融资额达到1.34亿美元的大笔融资出现。与此同时,器械巨头近年来的大手笔收购虽然加剧了竞争,但的确也对初创企业产生了巨大的激励作用。
相对巨头们的财大气粗,规模稍小的企业无疑只能通过其他的方式来补足手术机器人领域的不足,研发当然是手段之一。在Medical Design & Outsourcing网站公布的2019年全球最重视研发(研发费用占营收比例)的医疗器械企业TOP10中,研发费用占应收比例最高的TransEnterix便是一家腔镜手术机器人公司。
2019年,TransEnterix的研发费用为2182万美元。虽然绝对值不算高,但研发费用占其营收的比例高达90.5%——几乎可以认为除了运营必须的费用,这家企业所赚的每一分钱都被用到研发上去了。考虑到它只是在百强中刚刚上榜(排名98),再加上这一年来巨头进入这一领域导致的竞争加剧,大幅加强研发显然是一个合理的选择。
那么,既然达芬奇手术机器人已经占尽先机,为何大家还在这一领域上扎堆投入呢?
首先,手术机器人市场有着广阔的市场潜力。据市场咨询及调查机构MarketsandMarkets测算,全球医疗机器人市场规模在2027年有望突破221亿美元,相比2018年64.6亿的市场规模上涨3倍多。手术机器人在其中占据约60%的份额。随着全球人口老年化的加剧,心血管、神经血管和肿瘤等慢性疾病的发病率不断上升,未来的外科手术数量必然大幅增加,这给手术机器人带来更大的市场。
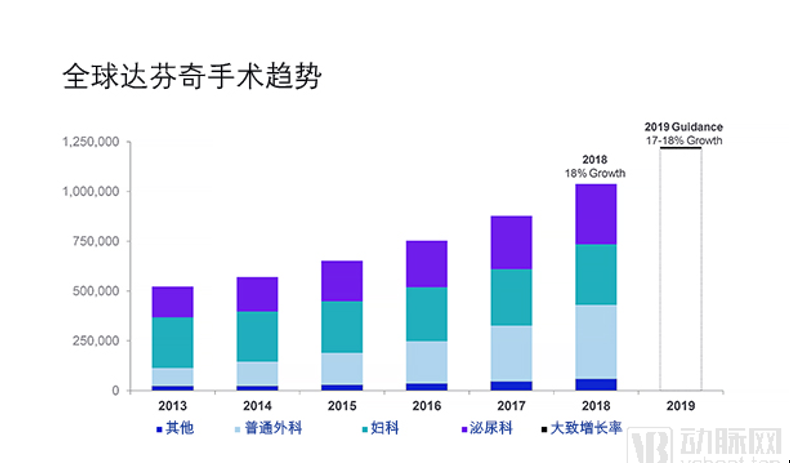
数据图片来自直观手术
其次,现有手术机器人的局限性使其在手术中所占比例并不高。达芬奇虽然声名在外,但价格高昂,单台价格2000万元左右。与此同时,达芬奇机器人的维护成本极为高昂,一年维护费大致在200万,且线驱动等耗材每使用10次就强制要求更换。这使其在全球部署的数量也不过5000台左右,且5-10万元的手术价格也并非普通人可以承受。
与此同时,达芬奇手术机器人也并非万能。作为典型的腔镜手术机器人,它在软组织手术上占据优势,但对于类似骨科等硬组织手术时就无能为力。
正因为此,根据统计,目前全球只有2%的手术是借助手术机器人完成。对于这些新入场的巨头而言,还未被发掘的98%的手术市场显然是一座亟待开发的金矿。
与此同时,达芬奇手术机器人为直观手术带来的超高收益足够让器械巨头们眼红。直观手术的营收年复合增长率高达45%,至2017年,直观手术实现营收31亿美元,同比增长15%。同时,自2005年后,这家公司的毛利率可以长年维持在70%左右,净利率一直高于20%,明显高于行业平均水平。
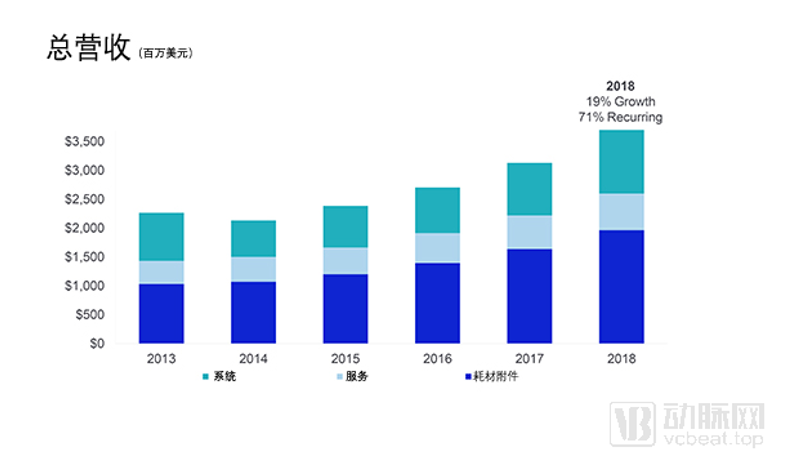
直观手术近年营收,数据图片来自直观手术
最后,器械巨头进入手术机器人领域还与自己的布局有关。目前,医疗器械设备向数字化和智能化转型的趋势十分明显。器械巨头通过收购领先的机器人公司快速入场,将其与自己的强势业务结合提供一体化解决方案。此举可以加强优势业务,并能够刺激现有业务并带来增长亮点。
随着众多巨头的入场,手术机器人市场的竞争也将会加剧,将有望迎来“百花齐放百家争鸣”的局面。这无疑是件好事,毕竟,良性竞争将会极大地刺激行业的发展。
手术机器人数字化智能化成趋势
目前的手术机器人存在体积过于庞大、成本高且人机界面不友好等主要缺点。目前,行业正在进行攻关,力图在这几个方面更进一步。从2019年的行业发展来看,手术机器人技术上呈现出下列趋势。
单孔机器人将取代多孔机器人
作为手术机器人的龙头企业,直观手术一直紧随技术趋势对达芬奇手术机器人进行改进。2018年,全新的单孔手术机器人达芬奇SP通过FDA认证,并很快在2019年通过了第二次FDA认证,显示了直观手术对其的重视程度。
不同于多孔手术机器人,达芬奇SP这样的单孔手术机器人只需要在病人身上打一个孔即可完成手术。因此,单孔手术机器人的机械臂具有6-7个自由度的灵活性。借由倍增的自由度,手术操作可以更灵巧更精细。
机械臂的减少使得单孔机器人体积更小,需要手术室空间要求更低。在安全性上,单孔手术机器人不会产生术中各机械臂的碰撞干涉,手术安全性更高。
单孔机器人可以避免多操作臂复杂的术前摆位,简化手术流程,进而减少病人麻醉时间。同时,它造成的创伤更小,病人术后恢复更快。从费用来说,单孔手术机器人的手术耗材更少,成本更低,更利于市场化。
5G+VR实现手术机器人远程操控,操作更真实
2019年2月,在西班牙巴塞罗那举办的MWC世界移动通信大会上,西班牙医生Antonio De Lacy在巴塞罗那会议中心通过5G视频连接到医院,对医院内患有肠道肿瘤的患者提供了远程手术指导。这是世界上第一台通过5G技术进行远程辅助的手术。
随后,2019年8月27日,5G远程应用更进一步。天津市第一中心医院骨科远程手术中心在中国电信天津分公司、华为技术有限公司的协助下,通过远程系统控制平台与北京积水潭医院连接,完成了国内首例骨科机器人5G远程手术。
这些事实都说明,5G与手术机器人的结合在2019年已经成为现实。远程手术需要超高清画面(分辨率至少4K)才能让医生看清画面并精确地进行操作。与此同时,远程手术需要同时连接生命监测仪、心电图机、除颤监护仪和高清视频设备等设备,连接主体数量较多且需要实时同步保证操作的安全性。这要求网络带宽达到15Mbps~1Gbps,延时在1ms左右才能满足相关操作要求。高带宽低延迟的5G完美满足了这一条件。
由于我国在5G上的强势地位,这无疑也将成为国产手术机器人弯道超车的一个绝佳机会。
与此同时,VR+手术机器人也在2019年崭露头角。2019年7月8日,国际着名微创手术专家、上海市微创外科临床医学中心主任郑民华在上海交通大学医学院附属瑞金医院胃肠外科/上海市微创外科临床医学中心为一名63岁女性患者实施了腹腔镜右半结肠癌根治手术。
这例手术特殊的地方在于它不单应用了5G技术,也将VR引入其中——在郑民华主任进行手术的同时,近20名学员戴上VR眼镜,或直接观看4K超高清大屏幕,腹腔内每一个细节都清晰呈现。这是国内首次实现5G+4K/8K+VR的腹腔镜手术直播。
尽管这例手术并未使用手术机器人,但将两者的结合无疑具有巨大的想象空间。
2019年完成融资的初创手术机器人企业中,的确有一家名为Vicarious Surgical的公司专长在这个方向。这家成立于2014年的公司就正在尝试将VR技术应用到微创外科手术中,开发一种专为微创手术设计的虚拟现实软件。
借助VR设备,医生可以实现对病人体内纳米手术机器人的控制,从而完成精细的微创手术。通过VR头盔透视病人的身体内部,医生能够更清晰地观察患者的病灶,从而提升手术效果。一旦遇上突发情况,医生也能够凭借虚拟现实技术更为便利地找到出现问题的原因,在微型手术机器人的配合下,降低手术的风险性,提高治愈率。
同时,VR技术还可以提升医生学习手术操作的效率。医生们不仅可以更为有效直观地观察到操作步骤,也可以亲身上阵,体验操作过程。并根据自身情况,及时调整学习的进程和范围,这种可谓理论与实践相结合的学习模式,将有机会大大提高医疗从业人员的培训进度。
在以往,网络延时对于VR来说是个巨大的挑战。随着5G的成熟,VR与手术机器人的结合算是扫清了一大障碍。
AI让手术机器人更智能
借助AI技术实现自动化手术导航功能将是机器人的发展方向。所谓自动化手术导航即是利用AI识别人体器官和手术机械,根据手术前的规划为外科医生进行术中指导,它可以规划手术路径,进行术中提示。
目前手术导航与手术机器人的结合主要应用在骨科,其核心要求是配准。骨骼属于人体硬质器官,空间比较固定,因此现阶段可以较为方便地应用手术导航。
利用AI对手术视频实时解析,手术机器人可以在手术中作出合理选择,比如规划下一步手术操作,或者通过各种方式预警主动脉等危险区域。同时,当医生在手术中遇到困难时,AI可以利用大数据给出建议,如调用专家的手术录像。在遇到极端场景时,AI将产生正面的帮助。
手术导航是未来自动化手术的一个技术基础,也是真正意义上机器人手术的必备功能。它可以降低手术难度,提高手术质量,同时可以辅助新外科医生的培训。
随着技术的进一步发展,自动化手术机器人将成为手术机器人的下一步目标。2016年,美国的STAR研究团队曾演示了STAR自动缝合机器人概念。这一概念产品通过全景3D摄像头和红外荧光成像自己规划缝合任务,并随着组织在手术中的移动变化不断调整自己的计划,在外科医生监督下实现了动物小肠自动缝合。
除此以外,Alphabet(谷歌母公司)旗下生命科学部门Verily与强生外科医疗器械部门Ethicon在2015年成立的Verb Surgical一直致力于研发智能外科手术平台,希望能够为外科领域带来颠覆式创新。
不同于其他手术机器人只聚焦于一个或几个环节,如术前规划、术中导航或对术中诊断有所创新,或解决医生手术技能提升的问题。Verb Surgical的目标是提供全流程手术AI解决方案。既通过高水平的底层AI操作系统(AI算法、数据),支撑术前、术中、术后不同的软硬件应用场景;又配置有多种多样的垂直治疗领域解决方案(智能器械、医用机器人、增强影像),以满足不同科室不同术式的应用需求。
如果能够完成这一愿景,将会是外科手术发生颠覆的一天。
国产手术机器人的2019突破
尽管在全球所占比例不高,但我国事实上对于手术机器人有着旺盛的需求。根据直觉外科公司公告统计的数据,我国每2000万人口所拥有的达芬奇手术机器人数量仅为1台,而美国及日本分别为147台及34台。2017年我国达芬奇年台均手术量高达388例,而同期世界平均水平仅为198例;2008年-2016年,我国手术总量8年CAGR为10.55%,同期达芬奇手术量年增长为84.48%,可见,我国的手术机器人市场空间极大。
达芬奇手术机器人极其高昂的费用是造成这一现象的主要原因。根据公开的数据,达芬奇手术系统(整机)的国内售价约2000万元;包含线驱动的机械操作臂作为耗材,每只限制使用10次,单臂更换需数万元。因此,国内使用达芬奇做手术,单笔费用至少在5万-10万元。
尽管成本高昂,但手术机器人所具备的扩大10倍视野、超越人手极限的精细稳定操作、大大减少出血量及并发症、大大缩短术后恢复时间等优点,又使得机器人手术的推广势在必行。
手术机器人的出现,使得顶级医生可以更快更好地完成手术,延长他们的职业寿命;也让年资较轻的医生可以大大提升手术质量。
手术机器人的国产化是解决这些矛盾的最佳解决方案。作为进口替代的一部分,我国在手术机器人的道路上一直奋起直追。在政策层面,国家陆续出台了多个战略规划和支持政策促进机器人产业的健康和快速发展,包括《中国制造2025》《机器人产业发展规划(2016—2020年)》等。
2018年11月,国家药品监督管理局修订了《创新医疗器械特别审查程序》,批准骨科手术机器人等21个创新医疗器械上市,降低临床治疗成本。
在这些政策的刺激支持下,国产机器人企业得到了长足的发展。
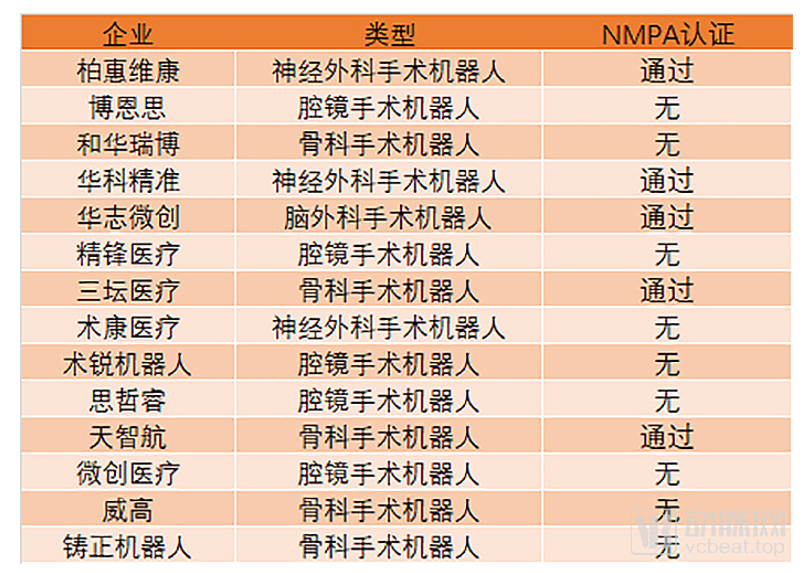
由于机器人的研发和生产周期长,成本高。因此,国内医疗机器人行业和其他科技医疗行业相比,在企业特点上有很大的差别。基本以大型企业为主,初创企业相对其他领域少了很多。同时,从事该领域的企业都有较大的资金投入,融资需求相对偏少。
尽管如此,在2019年,国内仍然有4家手术机器人企业获得了融资,分别对应了腔镜、神经外科和脊柱外科三类手术机器人。
在几类手术机器人之中,国产骨科机器人是最先获得突破的。2019年8月7号,上交所披露北京天智航医疗科技股份有限公司(简称:天智航)的科创板上市申请获受理。天智航的骨科手术机器人早已通过NMPA认证,以治疗设备及器械类唯一的“国际原创”产品入选了科技部《创新医疗器械产品目录(2018)》,并在多家医院临床应用。
根据天智航公开的科创板上市招股书披露的信息,天智航的骨科手术机器人从2017年11月首次部署在北京积水潭医院。迄今为止,其骨科手术机器人已经部署在20多个省/直辖市/自治区的50余家三甲医院,并已经累计完成超过3800例手术。
国产神经外科机器人在今年也获得了突破。2018年12月25日,华科精准研发的神经外科手术机器人正式通过国家药品监督管理局(NMPA)审批准产。这是首款获得国家创新审评通过的国产神经外科手术机器人,同时适用于儿童和成人。
另外,根据2019年9月8日国家神经外科手术机器人应用示范项目启动会暨学术交流研讨会上国家卫健委规划发展与信息司透露的信息,全国已经有10家大型三甲医院开展了柏惠维康“睿米”神经外科机器人辅助的DBS神经外科手术。
在达芬奇手术机器人占据优势的腔镜手术机器人领域,国产企业目前还没有获得认证。达芬奇处于垄断地位。
根据达芬奇在国内的独家代理商复星直观在2019年进博会期间公布的信息,达芬奇手术机器人目前在中国内地84家医院实现装机102台,在中国香港地区装机8台,累计手术量达12万例。在2018年进博会期间,复星直观透露的信息为内地装机70多台,香港地区10台,完成手术近10万例。
也就是说,短短一年的时间,达芬奇装机量增加了30台左右,完成手术2万多例。这也与复星医药2018财报中所提到“达芬奇手术机器人在中国内地及香港地区手术量同比增长超过20%”的说法相符。其中,泌尿外科手术超过四成。
从我国医院屡屡刷新达芬奇手术机器人单台手术量可以看出,我国达芬奇手术机器人代表的腔镜手术机器人有着极为旺盛的需求。浙江大学医学院附属第一医院先后在2015年和2016年刷新达芬奇手术机器人全球单台手术量。到了2018年,郑州大学第一附属医院以全年单台1198例手术创造了新的纪录。
尽管国产腔镜手术机器人还未获得认证,但包括微创医疗、思哲睿和精锋医疗在内的国产腔镜机器人目前已经进入到临床验证阶段,预计在2020年国产腔镜手术机器人就有望上市,从而打破达芬奇的垄断态势。
参考资料:
苏州智能制造:智能医疗|医疗机器人发展现状及未来五大趋势预测
贝壳社:深度报告|手术机器人行业调研
亿欧:手术机器人发展趋势:单孔手术机器人未来称王
硅兔赛跑:VR+手术机器人=华佗再世!
雷锋网:全军肝胆外科研究所刘荣:智能外科时代,手术机器人将由 AI 进行驱动 | CCF-GAIR 2019
器械之家:神秘的Verb Surgical手术4.0:“谁再说我是机器人公司我跟谁急!”
前瞻产业研究院:预见2019:《中国医疗机器人产业全景图谱》
来源:动脉网 作者:陈鹏


