






华为正式进军医疗器械
2021-08-11
日前,广东省药监局正式公布了《广东省医疗器械注册人制度试点批准产品名单》(截至2021-8-2),据了解,华为拿到了医疗器械注册证,产品名称为:腕部单导心电采集器,受托人为潍坊京为高科电子科技有限公司。

今日,企查查消息显示:华为终端有限公司所属集团是华为,注册资本为60000万人民币,成立日期为2012年11月23日,人员规模在10000人以上,从其经营范围来看,包括开发、生产、销售:医疗器械(第一类、第二类、第三类医疗器械),并提供技术咨询和售后服务;增值电信业务经营;佣金代理;货物或技术进出口(国家禁止或涉及行政审批的货物和技术进出口除外)。

今年5月,华为消费者业务手机产品线总裁“何刚”微博发文指出,华为首款可以测量血压的智能手表已通过医疗器械注册检验,下一步将联合专业医疗机构开启注册临床试验,预计在下半年将正式上市。
何刚表示,一直以来,华为在健康领域持续深耕,致力于为大家带来更加专业的健康服务。此前何刚在花粉年会上透露,华为正在研发可穿戴血压测量设备,并将与国内知名机构启动高血压管理研究,探索从筛查到早期干预的血压创新主动健康管理。
华为还突破了基于智能穿戴的体温连续检测技术,正在开展智能体温的联合研究,实现体温异常筛查。华为正在进行冠心病智能筛查与预警的联合研究,借助华为智能手表心电采集能力,融合 PPG、ACC 数据实现冠心病智能筛查与预警,帮助消费者主动管理健康。
心电监测是第一落点?
今年2月,据企查查显示,华为技术有限公司关于 “用于心电测量的装置和方法”专利信息被授权公开,专利公开号为 CN108601544B。发明实施例涉及医疗器械技术领域,并且更具体地,涉及用于心电测量的装置和方法。
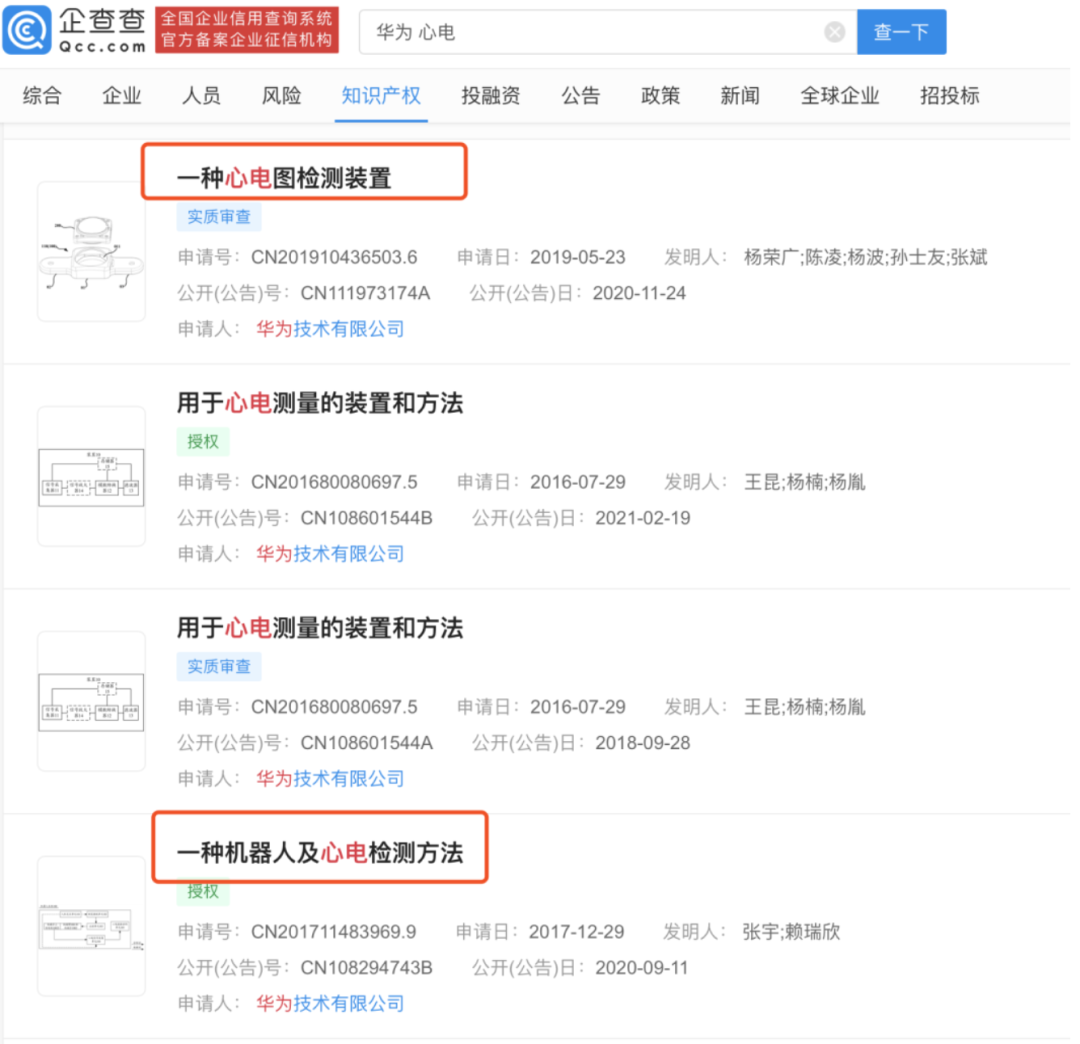
这项专利于 2016 年 7 月 29 日开始申请。专利描述了使用多个电势传感器捕捉电信号,然后通过信号放大器、模数转换器、滤波器等进行信号处理,从而获得有效心电信号的方法。
据了解, 本项专利的申请人为:华为技术有限公司;申请人地址为广东省深圳市龙岗区坂田华为总部办公楼。
华为还在2020年11月24日公开了一种心电图检测装置,该申请提供了一种心电图检测装置,以降低心电图检测装置的设计与制造难度。
据悉,该心电图检测装置包括可穿戴本体以及心电图检测模组,其中:心电图检测模组与可穿戴本体可拆卸连接,心电图检测模组包括壳体、基板、心电采集电路以及内电极和外电极,基板设置于壳体内,心电采集电路设置于基板上,内电极和外电极设置于壳体的外壁,且内电极靠近基板的一侧设置并与心电采集电路电连接,外电极靠近基板的另一侧设置并与心电采集电路电连接。
华为在2020年9月公开了一种机器人及心电检测方法。解决现有采用便携式心电记录仪心电检测时操作不便的问题。
在医疗领域争一杯羹
作为国内科技企业的代表,华为一直在医疗领域布局。
华为官方显示,华为推出了:华为Atlas新冠肺炎智能评价解决方案,Atlas人工智能计算平台,与医疗PACS设备相结合,使之具有边缘AI计算能力,可以做到实时病灶多色渲染、肺炎自动分级、基线直方图直观对比,患者肺炎评估效率相比传统方案提升9倍以上。

2020年4月1日,华为投资的荣耀终端有限公司成立,注册资本3亿人民币。该公司由华为投资控股有限公司全资持股,华为消费者业务CEO余承东担任公司董事长。
2019年11月末,华为旗下全资子公司哈勃科技投资有限公司投资的深思考人工智能机器人科技(北京)有限公司经营业务发生变更,销售医疗器械I、II类、计算机、软件及复制设备。
2019年3月末,华为变更公司经营范围,新增医疗器械销售等业务。华为终端有限公司变更公司经营范围,新增了医疗器械(第二类医疗器械)销售等业务。
针对入局医疗器械市场,据每日经济新闻报道,华为方面回应称,华为会做与医疗设备连接的设备;在这一基础上,未来可能会和合作者为市场提供健康医疗辅助解决方案。
与此同时华为官网还显示:华为医疗目前已经服务超过3000家医疗卫生服务机构。为医疗行业提供全面覆盖,安全可控,运维简单,面向未来的无线网络解决方案。
据动脉网报道,在郑大附一院的建设案例中,华为Wireless XLabs负责人曾表示,华为与运营商一起合作为客户提供5G解决方案。据悉,第一台远程5G动物手术以及人体手术就是由华为分别和联通、移动合作完成的。 同时,华为在无线应用场景实验室Xlabs的探索过程中,已经在多个方向上有了进展,其中包含运营商、医院、医疗设备商等产业链280多个合作伙伴,形成了40多个合作研究项目。
在信息化建设上,华为还曾服务过一大批三甲医院。 与华西医院共同建设了大数据集成与应用平台,和客户一起规划设计了华西二院新院区数字化医院顶层设计和实施。与同仁医院构建安防保障体系;用微模块解决方案帮助中国人民解放军总医院、南京鼓楼等医院建设了高集成、易维护的数据中心。
华为未来还有哪些动作,让我们拭目以待。
来源:医械世界


