






微创医疗机器人今日在港上市
2021-11-04
11月2日,港交所迎来首家手术机器人企业——微创医疗机器人。截至今日上午十点,微创医疗机器人报每股43.2港元,市值超400亿港元。
3款旗舰产品纳入创新医疗器械绿色通道,其中1款上市
微创医疗机器人成立于2015年,为微创医疗集团子公司,专注于手术机器人的设计、开发及商业化,根据弗若斯特沙利文的资料,该公司是全球行业中唯一一家拥有覆盖五大主要和快速增长的手术专科(即腔镜、骨科、泛血管、经自然腔道及经皮穿刺手术)产品组合的公司。
目前微创医疗机器人已推出三款旗舰产品——图迈腔镜手术机器人、蜻蜓眼三维电子腹腔内窥镜、鸿鹄骨科手术机器人,三款产品均已被纳入国家药监局的创新医疗器械特别审查程序,目前图迈及鸿鹄处于注册批准阶段,而蜻蜓眼已于今年6月获得国家药监局批准,成为首批由中国企业开发的商业化三维电子腹腔内窥镜。
图迈由患者手术平台、图像台车、医生控制台三部分组成,用于辅助完成腔镜外科手术,特别是对于开放术式或常规腹腔镜术式具有挑战的复杂手术,具有明显的优势。目前图迈已完成应用于泌尿外科手术的注册临床试验并递交注册申请,成为首款亦是截至目前唯一一款由中国企业研发、完成注册临床试验并递交注册申请的四臂腔镜手术机器人。

在一项前瞻性、多中心、随机及平行对照试验中的临床结果显示,通过与达芬奇 Si手术系统的比较评估,图迈用于泌尿外科手术的有效性及安全性不劣于达芬奇Si ,并且几乎所有次要有效性终点均无统计学显着差异。
蜻蜓眼包括三维电子腹腔内窥镜及图像处理器,为检查腹部、胸腔及骨盆区等器官而设计。与传统二维腹腔镜相比,蜻蜓眼通过双路图像采集的方式,为术者提供手术视野的三维立体感和手术操作的纵深感,为术者在腹腔镜下完成快速精细定向操作,如手工缝合、吻合及功能重建、持针器械的换手、打结等提供非常重要的帮助,进一步提高手术的安全性。

骨科手术机器人鸿鹄是一款用于辅助关节置换的手术机器人,也是截至目前唯一一款由中国企业研发且配备自主研发机械臂的关节置换手术机器人。其术前规划系统可根据患者术前CT扫描数据建立膝关节三维模型,根据患者生理解剖学特征生成个性化假体植入手术方案。目前鸿鹄已完成应用于辅助膝关节置换手术的临床试验并递交注册申请。

除旗舰产品外,微创医疗机器人还有六款产品在研。

尚处亏损状态
财务方面,微创医疗机器人目前尚无盈利。截至2019年及2020年12月31日止年度以及截至2020年及2021年6月30日止六个月,其分别录得亏损净额6980.1万元、约2.093亿元、约4896万元及2.426亿元,主要为研发投入和行政开支。
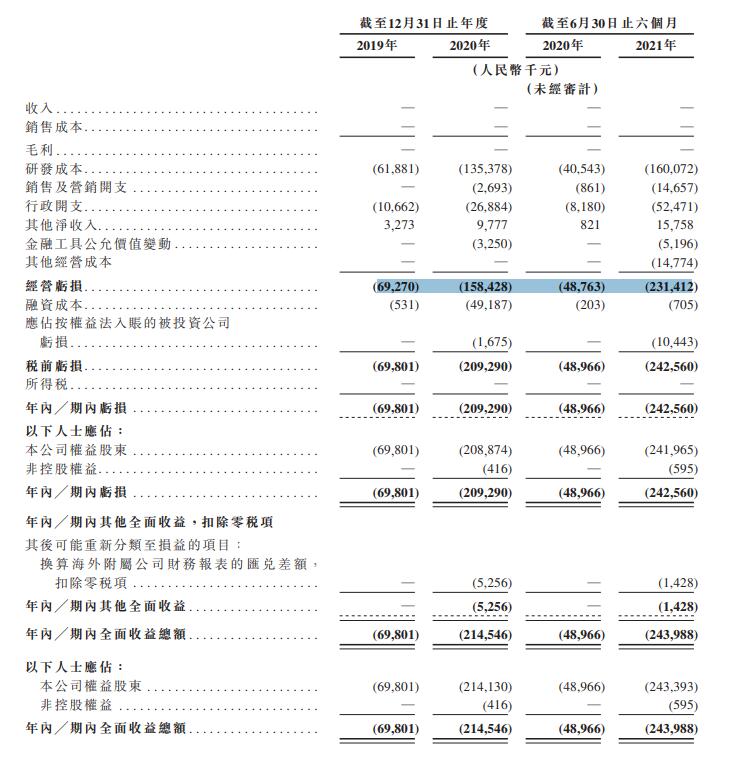
研发投入方面,财务数据显示截至2020年6月30日止六个月的人民币4050万元大幅增加至截至2021年6月30日止六个月的人民币1.601亿元。研发成本分别占经营开支总额的85.3%、82.1%、81.8%及 70.5%。微创医疗机器人方面称,研发投入增加的主要原因系图迈及鸿鹄的注册临床试验取得进展及其他在研产品的开发,导致研发雇员数目增加及材料及消耗品成本增加。
国内手术机器人市场加速发展
近年来,全球手术机器人市场蓬勃发展,国内手术机器人市场目前虽处于早期发展阶段,但增长潜力巨大。2020年,中国手术机器人市场的市场规模为425.3百万美元(约27亿人民币)。预期手术机器人市场将以44.3%的复合年增长率快速增长,2026年达到3840百万美元。
目前国内手术机器人企业包括天智航、微创医疗机器人、山东威高、键嘉机器人、三坛医疗、鑫君特、罗森博特等,其中天智航于2020年7月登陆科创板,成为“国产手术机器人第一股”。天智航主要专注于骨科手术机器人领域,2016年其骨科手术机器人“天玑1.0”获批上市,作为通用型骨科手术导航定位机器人产品,“天玑1.0”能够覆盖骨盆、髋臼、四肢等部位的创伤手术及全节段脊柱外科手术。天智航近年来的主营业务收入也主要来自于“天玑1.0”的销售,财报显示,天智航2017-2020年的营收分别为7329万元、1.27亿元、2.3亿元以及1.36亿元。今年4月,产品功能与结构均进一步优化的“天玑2.0”骨科手术机器人国内版获得NMPA核发的第三类医疗器械注册证,国际版完成样机开发,已通过CSA获得IEC60601-1医疗电气设备-基本安全和主要性能通用要求的认证。
同时,10月27日山东威高的“腹腔内窥镜手术设备”获国家药监局批准上市,成为国内首家获批的腔镜手术机器人。长期以来,国内腔镜手术机器人市场基本为美国达芬奇手术机器人占据,而一台“达芬奇”机器人手术设备售价高达2000多万,高额的售价以及维护、耗材成本,给医疗机构和患者就医带来了较大的负担。作为国内首台具有自主知识产权的手术机器人,山东威高的“腹腔内窥镜手术设备”攻克了被国外垄断了20年的技术难题。2020年9月24日,由青岛大学附属医院副院长牛海涛领衔的手术团队成功跨越3000多公里的空间障碍,利用5G远程技术,为贵州西秀区人民医院手术室内的71岁男性膀胱癌患者成功手术。这是世界上首例5G超远程自主原研机器人辅助腹腔镜手术。
值得一提的是,今年4月上海医保局将28个新项目纳入上海市基本医保支付范围,其中“人工智能辅助治疗技术”即与达芬奇手术机器人有关。目前“达芬奇手术机器人”医保报销范围包括前列腺癌根治术、肾部分切除术、子宫全切术和直肠癌根治术4种手术。根据上海医保规定,达芬奇手术机器人支付类别为乙类,患者自负比例为20%。
同时,达芬奇手术机器人在国内也在不断加强本土化布局,今年10月,直观复星达芬奇创新中心在张江科学城上海国际医学园区开幕,总面积1700平方米,总投资超过1亿元,中心配备行业前沿的模拟培训设施和设备,提供高度仿真的培训环境和阶梯式专业临床培训,对国内医护专业人士开展达芬奇手术机器人试驾、技术培训及由临床专家带教的高级临床培训课程操练等项目,同时,其在张江科学城上海国际医学园区规划的“全球第二总部”也在加速推进中,未来将打造成为亚太地区达芬奇机器人与肺科机器人研发、生产、物流和临床教育的基地。
来源:医谷网


