






“翻盘”的国产血气分析
2021-01-22
2019年,国内的血气市场份额约为16.5亿元,随着疫情的爆发,血气的需求预计2020年增长将超过30%-50%。由于血气设备技术壁垒非常高,2013年之前,长期处于被进口垄断的局面,国产设备并未能撼动进口的地位。而在这次疫情期间,许多血气厂家反应迅速,在第一时间组织企业复工复产,积极参与到全国抗疫大战。国内疫情稳定之后,又迅速调整战略,出海世界上其他疫情国家。初略统计,部分厂家血气产品单个季度的业绩抵得上往年一年的收入!
本文,我们就一起来认识下因疫情“翻盘”的血气分析:
一、什么是血气分析
血气分析是危重症患者必不可少的检查手段,可用来判断机体是否存在酸碱平衡失调、判断通气换气功能、缺氧及缺氧程度等等。在急性呼吸衰竭诊疗、危重症监测、新生儿监测、心脑血管疾病、麻醉手术等过程中发挥着至关重要的作用。
二、血气分析的临床应用
对于血气分析技术,由于检测参数的特殊性,血气分析要求样本在采出的最短时间内得到测定,从而帮助临床医生进行快速准确地诊断并能及时有效地采取治疗措施。

从临床使用情况来看,进行血气分析检查的对象主要是老年患者、有基础疾病患者、肺功能不好的患者,以及对处于危急重的休克、昏迷、意识障碍、呼吸困难、胸痛、手术前中后的患者的监测等等。目前使用血气设备较多的以ICU、麻醉、急诊等科室患者为主。
ICU
ICU中的危重患者因机体内环境紊乱,常伴有多脏器功能损害,特别是肺和肾功能障碍,易并发动脉血气异常和酸碱平衡紊乱。因此及时正确地识别和处理对挽救重症患者的治疗更有指导作用。
麻醉患者
麻醉患者由于疾病、麻醉、手术以及术中出血和输血、输液的影响,很容易出现血气变化和酸碱失衡,而发生在麻醉中和麻醉恢复期间的心搏骤停约有60%与低氧血症和高碳酸血症有关。这一期间血气分析仪的应用能全面了解患者的呼吸功能,及时发现和准确诊断低氧血症与高碳酸血症,为正确处理麻醉患者所出现的血气变化和酸碱失衡提供依据。
急诊患者
临床上急诊科室的重症患者均可引起血气及电解质紊乱,导致患者酸碱失衡,而严重酸碱平衡紊乱又可加重对人体各脏器功能损害,有时可直接成为患者致死的原因,一份准确的血气分析结果尤为重要,可以有效帮助急诊科医生快速了解患者酸碱失衡情况并及时处理。
三、血气分析对新型冠状肺炎诊疗方案的制定具有指导意义
在《新型冠状病毒感染的肺炎诊疗方案(试行第七版)》中,明确表示了血气分析对本次新型冠状病毒肺炎诊疗方案的制定有着非常重要的指导意义。
血气分析是指对溶解于血液中的气体成分(O2、CO2等)的含量进行测定以及对酸碱平衡的有关指标进行测量,了解患者的通气功能,呼吸衰竭与严重程度,以及各类型的酸碱失衡状态。而血气分析是临床上非常重要的评估利器,它作为动态判断肺部通气和换气状态,及了解机体酸碱平衡情况的检测设备,在新冠肺炎重症患者诊疗中发挥着巨大作用,是一种常见的急诊、重症、抢救设备。如果没有它,ICU的医生相当于失去了一双洞察机体内环境的慧眼。

四、国产血气厂家
根据国家药监局官网查询到的数据,目前已获得血气分析产品注册证的国内厂家主要有深圳理邦、广州万孚、武汉明德、南京普朗、梅州康立等厂家。
以配套使用的试剂类别来区分,主要分为干式血气分析仪和湿式血气分析仪。以设备的外形区别,可分为便携式血气生化分析仪和非便携式血气分析仪。
此次疫情使得各级医疗单位充分意识到了POCT血气检测的重要性,许多POCT厂家的市场占有率和品牌影响力在此次疫情后得到了迅速提升。
五.《公共卫生防控救治能力建设方案》落地,血气分析进入后防疫时期急救“清单”
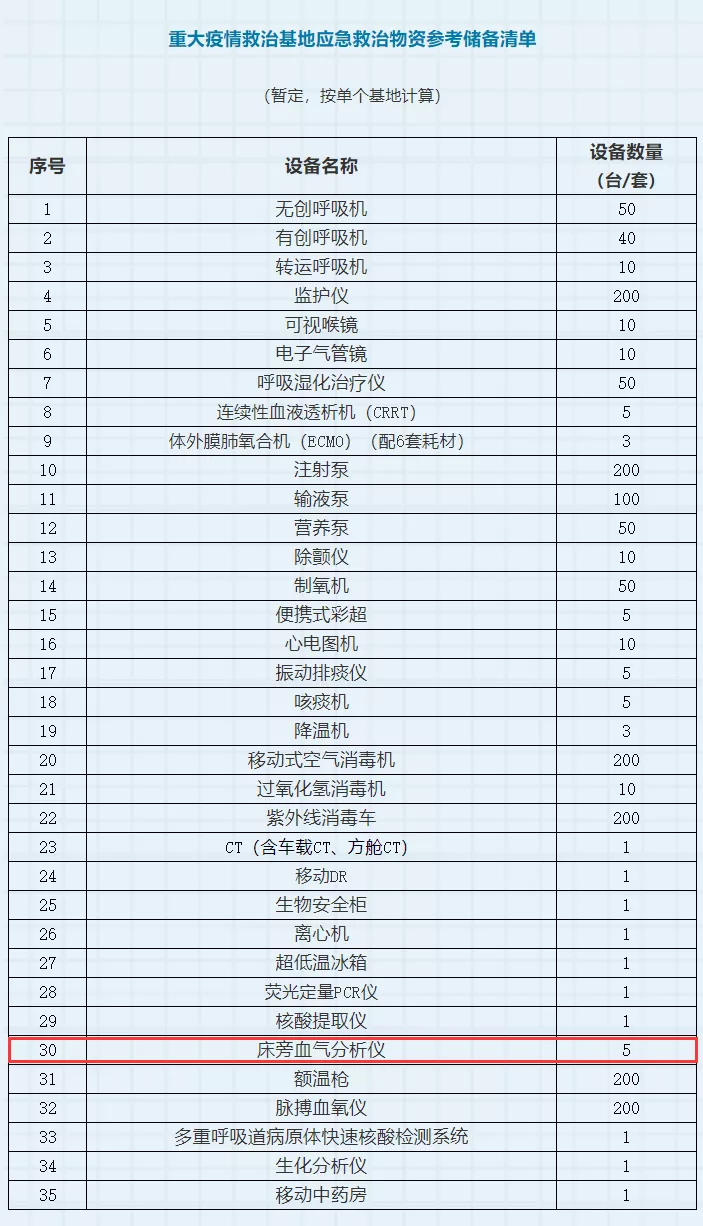
受此次疫情影响,上海公共卫生系统的专家们认为血气分析对新冠患者的早期诊断、监测治疗能起到重要作用。同时,在国家发展改革委推出的《公共卫生防控救治能力建设方案》中,床旁血气分析仪被列入到“重大疫情救治基地应急救治物资参考储备清单”中,按单个基地来推算,每个基地需要配置5台床旁血气分析仪。可见,经此一“疫”,国家非常重视血气设备的临床应用价值。
文章及图片来源:IVD资讯


