






效率能翻番,“叠层模具”技术你了解多少?
2021-10-08
01什么是叠层模具
叠层模具是当今塑料模具发展的一项前沿技术,型腔是分布在2个或多个层面上的,呈重叠式排列。简单地说,叠层模具就相当于将多副单层模具叠放在一起,安装在一台注塑机上进行注塑生产。
叠层模具产生的需求背景通常注塑机在使用单层塑料注射模具的时候,其本身的 注 射 量 和 开 模 行 程 只 使 用 了 额 定 的20%~40%,没有充分发挥注射机的性能,而叠层式注塑模具能够在不增加锁模力的基础上,在 1 台注射机 1 个操作人员的条件下能使产量根据层数成倍增加,可以极大地提高生产效率和设备利用率,并能节约生产成本和人力资源。叠层式注塑模具最适于成型大型扁平制件、浅腔壳体类制件、小型多腔薄壁制件和需大批量生产的制件。
02叠层模具的优点是什么
1)与常规模具相比,叠层式模具锁模力只提高了10%~15%,但产量可以增加90%~95%;
2)模具制造要求基本上与常规模具相同,且将多副型腔组合在一副模具中,所以模具制造周期也大大缩短;
3)使用高效叠层注塑模可以得到双倍或者四倍于普通单层模的产出而无须投资购买额外的机器和设备。这就节约了安装机器、设备和扩建厂房、新增劳动力的费用;
4)叠层模具可以装在单层模具要求相近的注塑机上,单位时间的产出效率却是成倍的增长
5)节约原料,易于自动化,提高产品性能,缩短生产周期
03叠层模具的分类
1)普通叠层模具
2层模
3层模
4层模
最多到10层
2)旋转叠层模具
双层双面每次180度旋转
双层4面每次90度旋转
3层2面(每次180度旋转)+4 面(每次90度旋转)
3)Tandem模

04叠层模具的发展历程
1)叠层式模具的原始模型是1899年美国人Alanson cD.Gray在铸造薄壁蜡制品时采用;
2)早在1940年12月E.R.Knowles就取得了叠层式模具的专利权,但未推广使用;
3)20世纪 60 年代瑞士的模具制造商开始研究用于加工塑料的叠层式模具,主要是生产包装盒及简单的日常用品的注塑模具;
4)1980 年德国人设计了普通流道的双层注射模(即冷流道叠层式模具);
5)20世纪80年代末90年代初日本人、德国人设计发明了各自不同结构的热流道叠层式模具;
6)20世纪90年代初Tradesco 模具公司的 Rozema研发出4层模;
7)21世纪初Tradesco模具公司成功开发了3层多腔叠层式热流道模具;
8)1995 年俄罗斯的 Grabovski研发出直角进浇热流道叠层式模具;
9)21世纪初德国人设计了一种16+16 腔的 2 种材料的旋转叠层式模具,之后用于多色注塑的旋转叠层模具逐渐广泛使用;
10)2002年由德国Bielefeld大学应用科学研究院开发了应用于厚壁制品的Tandem模具。
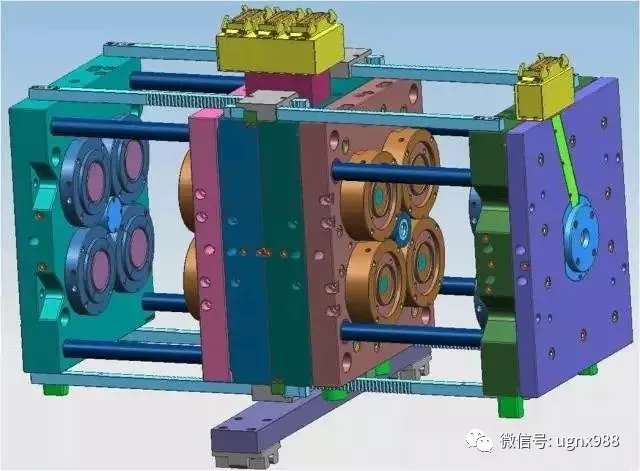
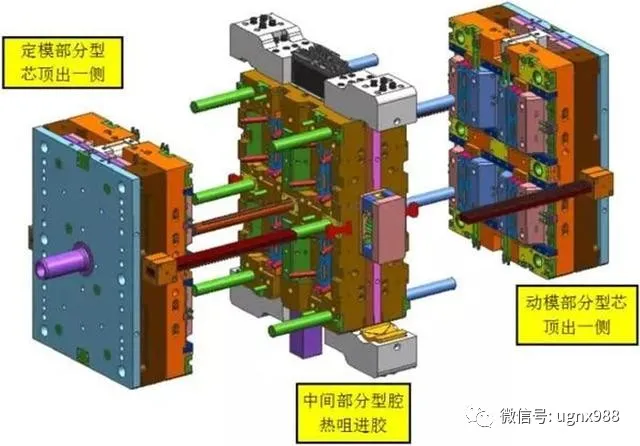
05叠层模具结构
2层叠层模具基本构成部分
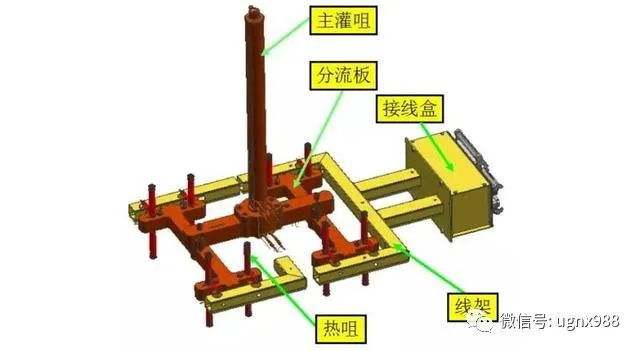
叠式热流道设计
1)主灌咀的运动导向
2)主灌咀与流道板的连接
3)流道板与热咀的连接
4)线架、接线盒与热流道部分的整体化设计
5)热流道的平衡技术
6)热流道装拆方便性考虑
7)热流道系统的保护
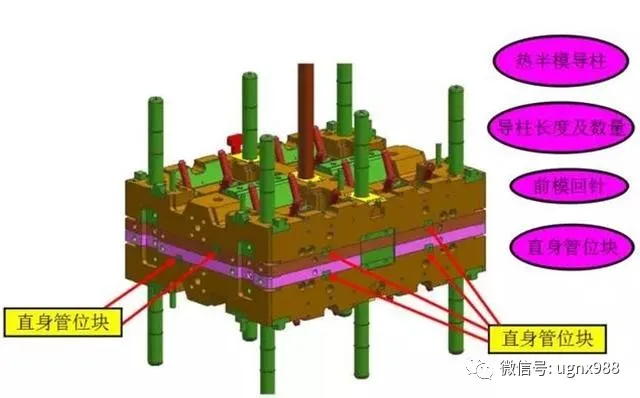
热咀保护
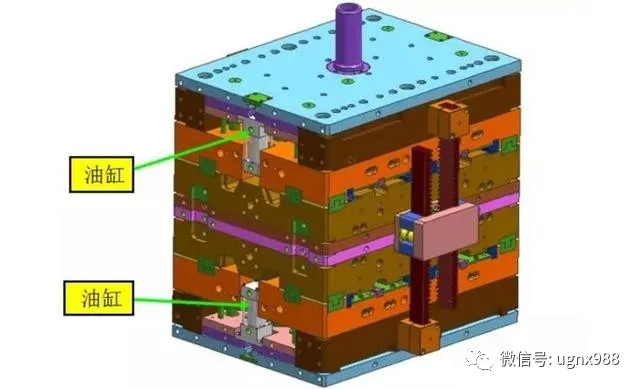
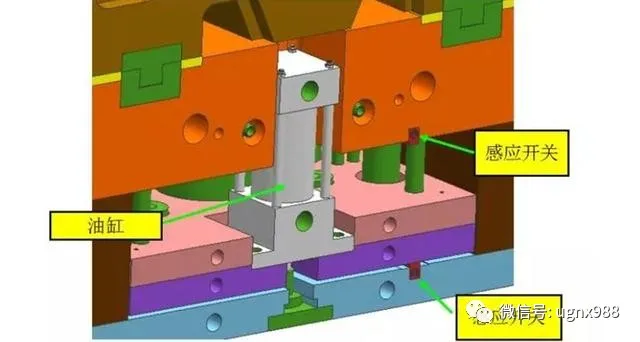
同步开模机构
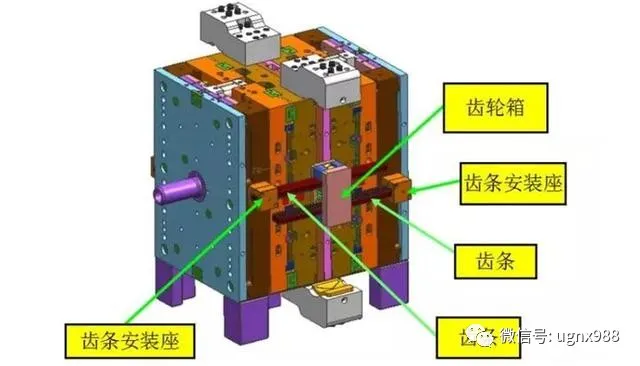
中间支撑导滑机构
顶出机构
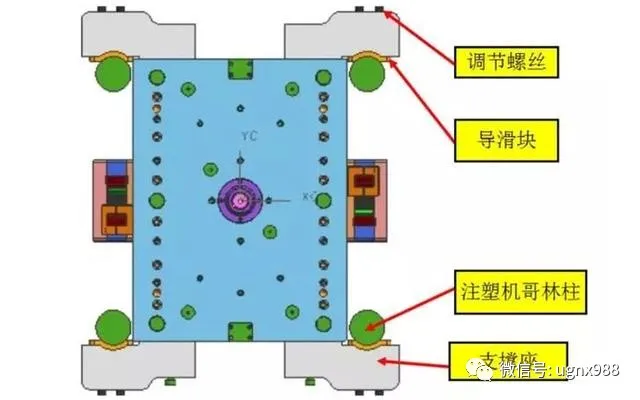
导向定位系统-模胚部分
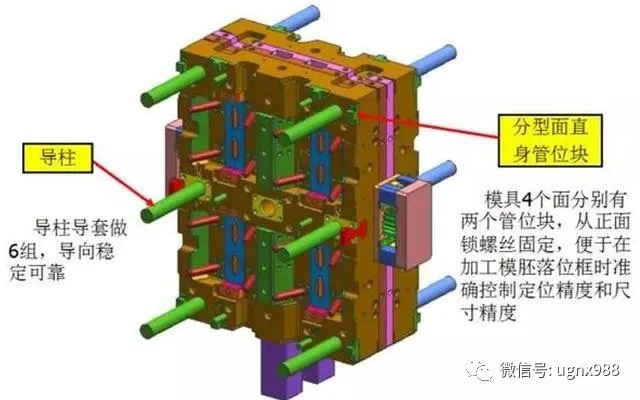
06叠层模具设计考虑要点
2)需校核注塑机最大开模行程和最大塑化量;
3)注射装置的预塑效率要高;
4)注射速率适当增加;
5)热流道熔体的压力释放;
6)型腔布置与主灌咀的关系处理。
来源:UG模具设计资料


