






2023上海医疗器械创新展政策法规演变之FDA伴随诊断监管
2023-09-12
2023上海医疗器械创新展Medtec 创新展分享本次报告《FDA伴随诊断监管政策的演变与案例研究》(Tracing the Evolution of FDA CDx Approval Policies Through Case Studies)的报告。胡云富博士为前美国FDA资深注册评审专家,在FDA任职11年期间,曾带领团队完成诸多里程碑式的伴随诊断(CDx)产品和服务审批工作。从FDA监管CDx的基本原则、CDx监管政策的演变历程、美国LDT和CDx监管的现状与未来三个方面进行了分享,将陆续分三期内容进行介绍。首先简单为大家介绍FDA监管医疗器械的法学基础。
01
FDA监管医疗器械的法学基础
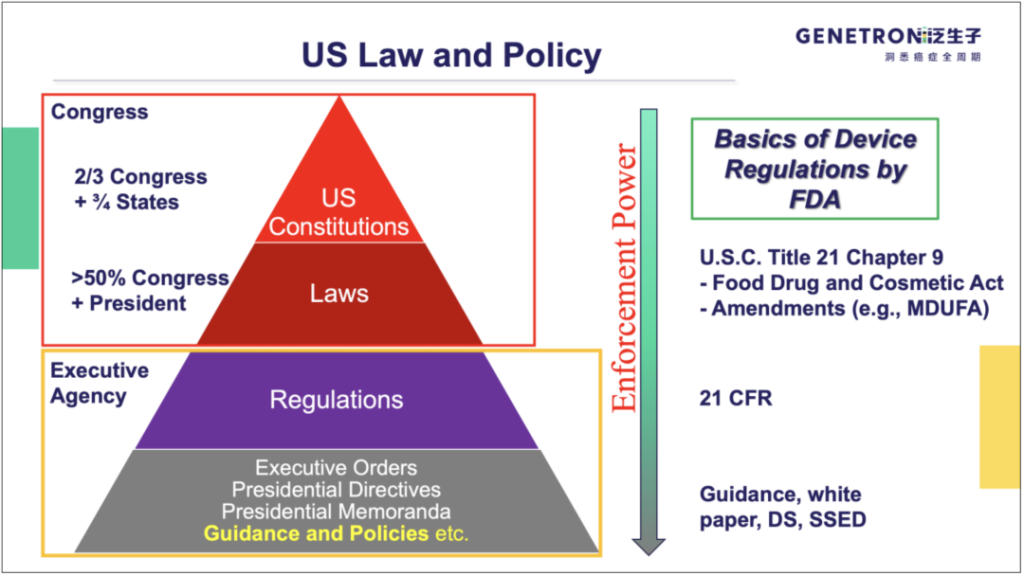
FDA监管医疗器械的法律基础是 21 U.S. Code Chapter 9(也被称为联邦食品、药品和化妆品法,FD&C Act)及其修正案。在法规层面,FDA发布的法规编纂于《Code of Federal Regulations》(CFR)第21章,可以在http://www.ecfr.gov/通过章节号或文本进行查询。在法规以下,FDA通常通过指南、白皮书、医疗器械获得许可/批准时的DS(Decision Summary)或SSED(Summary of Safety and Effectiveness Data)文件等,阐释其对医疗器械的具体监管要求和实施方式。值得指出的是,由于美国是判例法国家,既往CDx的评审案例及相应DS、SSED文件,对于后续CDx产品的申报具有重要参考意义。
02
CDx的监管
伴随诊断的概念曾于2006年见于文献[1],到2011年FDA发布了全球第一个体外伴随诊断设备指南(草案)[2]并在2014年形成正式版[3]。该指南将CDx定义成:为相应药物的安全、有效使用提供必不可少信息的体外诊断(IVD)。简单来说,CDx通过4个“合适”使患者获益,即在合适的时间为合适的患者在合适的剂量下使用合适的药物。
在实际的伴随诊断产品开发中,有时由于条件受限,主要药效临床试验使用的检测试剂可能并非是最终要进行申报的IVD产品,此时就会涉及桥接试验的开展。
医疗器械的安全可靠对全世界人类都至关重要,各国监管部门对医疗器械全生命周期质量风险管理都越来越严格,上市后监管也是各大企业需要面临的重要任务。按照我国新法规和规章的要求,“十四五”重点监测工作首次提出以注册人为重点的监测主体,从任务分工、工作机制上落实企业主体责任。2023上海医疗器械创新展Medtec 创新展将于年底2023年12-13日开展,同期举办质量:中外医疗器械质量论坛,以新视角切入医疗器械质量管理。
03
桥接研究(Bridging Study)
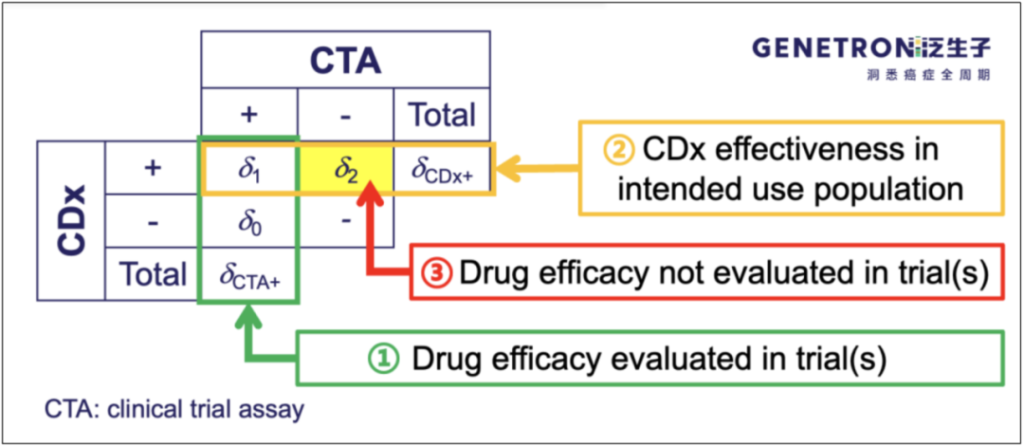
在实践中,比较常见的问题是CTA样本无法100%用来重测,特别是CTA阴性样本可能存在未入组/未签知情同意书等问题。如果大家阅读既往经桥接获批的CDx SSED文件,便会知道FDA允许采用药物临床试验以外的样本支持一致性研究。企业在药物临床时,应尽量确保知情同意允许重测并尽量保存阳性和阴性样本;遇到在阴性样本不足的情况,可以与FDA协商采用部分外部样本,但需要注意补充的外部阴性样本能否代表伴随诊断预期使用人群;阴性样本的数量必须结合突变的发生率、检测的特异性等用统计方法分析确定。
04
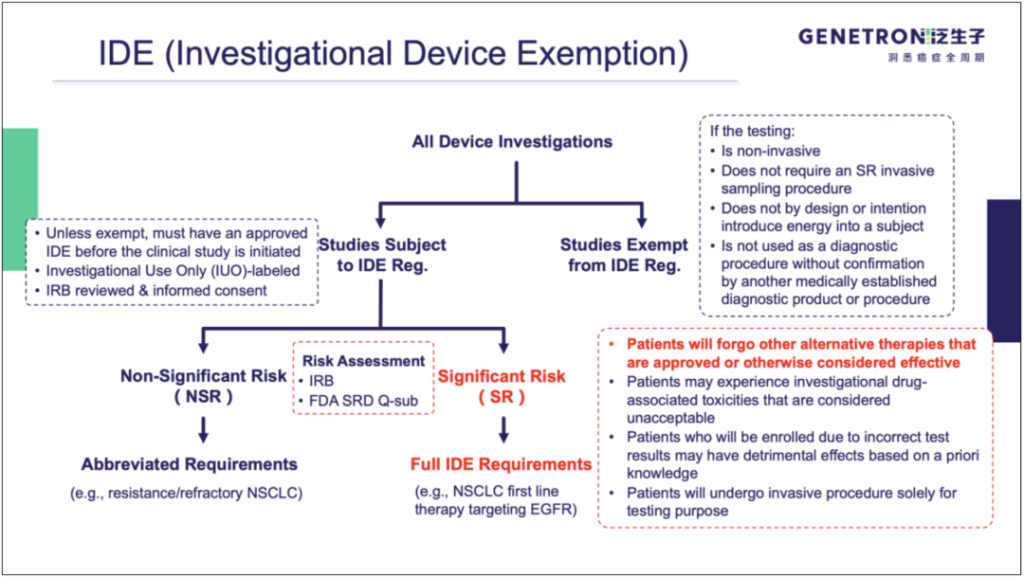
IDE与简化提交流程

Form FDA 1571
案例一
最早获批的伴随诊断产品——HerceptTest
1998年,FDA批准了第一个伴随诊断产品HerceptTest,用于抗乳腺癌药物赫赛汀的伴随诊断检测。此时,FDA还没有CDx概念和相应的审评政策,HerceptTest通过与CTA进行一致性研究的方式获批上市。
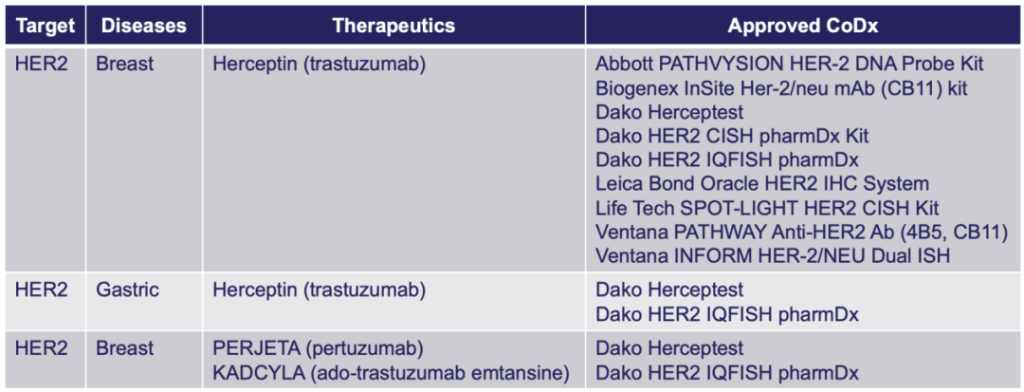
FDA批准的HER2 CDx[1]
案例二
监管科学与技术的共同进步——BRACAnalysis CDx
2014年,FDA批准的BRCA1/2基因检测——BRACAnalysis CDx也是极具代表意义的案例。这不仅是FDA批准的首例基于LDT(Laboratory Developed Tests)的CDx服务,而且是首个根据基因水平结果判定是否预期获益的CDx。在此之前,检测DNA的CDx都需要具体到基因的某个或某些变异。BRACAnalysis CDx审批过程中关注的是针对各类变异制定的分类标准;获批上市后,随着认知加深,分类标准可能会发生改变,申报方需要持续向FDA同步分类标准的变更。
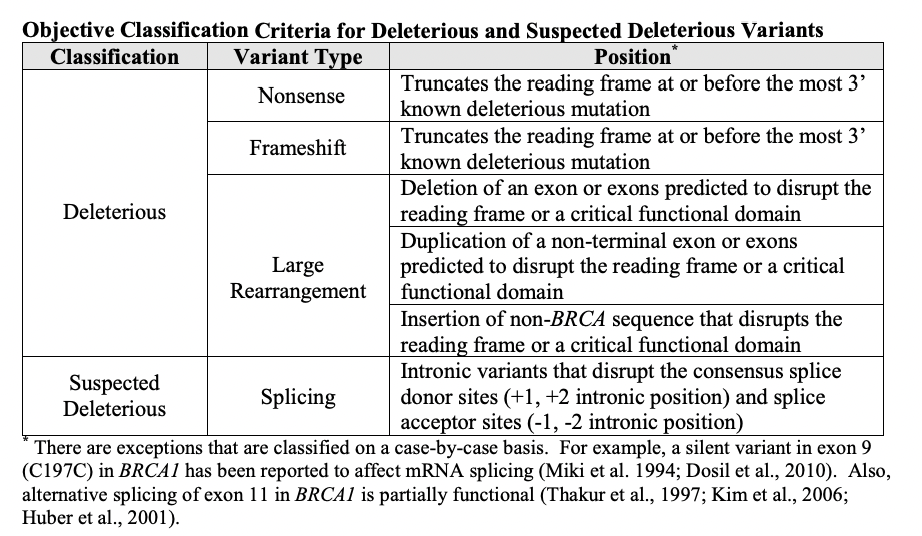
BRACAnalysis CDx原始PMA SSED中
关于变异分类标准的描述[2]
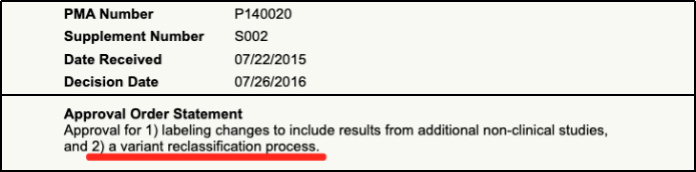
BRACAnalysis CDx对变异分类标准
进行调整的PMA补充记录[3]
此外,由于BRCA1/2胚系变异与乳腺癌和卵巢癌的家族遗传风险相关,一部分人群在未患病时就需要做检测,后续如果患病无需重复检测。相比此前FDA批准的检测体系变异的CDx,这是一种新的应用场景。为此,FDA将BRACAnalysis CDx预期用途中相关描述由“符合条件接受Lynparza治疗者” 调整为“符合条件或未来可能符合条件接受Lynparza治疗者”,从而拓宽了应用场景并支持了医保报销覆盖。预期用途中看似只有“几字之差”的变化,实际可以为患者来带更多获益。

案例三
审批范式的变迁——EGFR 突变检测
EGFR是FDA批准CDx种类特别多、审批流程也相对成熟的生物标志物。
01
原研CDx——桥接审批策略
2013年,FDA批准了首个EGFR突变CDx检测,cobas EGFR Mutation Test。支持其审批的主要研究通过桥接完成,包括与CTA的一致性研究以及临床有效性研究。此方法已成为目前非常常见的原研CDx审批策略,桥接介绍见上篇。
02
液体活检产品
2016年,FDA批准了第一款液体活检CDx——cobas EGFR Mutation Test v2。从其主要一致性研究数据可以看出,一部分组织样本检测阳性的患者,其ctDNA检测结果为阴性。这意味着虽然该产品的阳性结果可信,但其阴性结果不能反映受检者病灶不存在EGFR相应突变。因此,FDA要求cobas EGFR Mutation Test v2在预期用途中明确指出“检测结果为阴性的患者应进一步进行常规活检,并使用FFPE样本类型进行EGFR突变的检测”。对于后来获批的液体活检产品Guardant360 CDx,FDA也有相同要求。
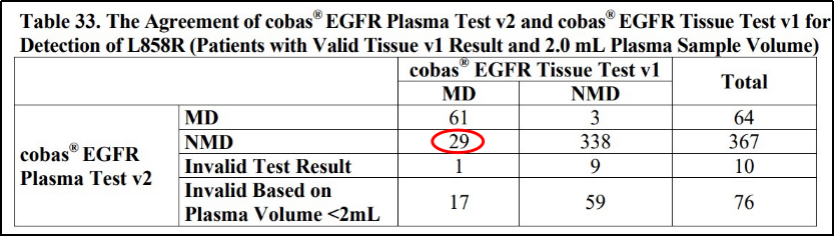
cobas EGFR Mutation Test v2液体活检一致性研究
部分结果及预期用途[4]
03Follow-on CDx——非劣效研究审批策略
2017年,FoundationOne CDx(F1CDx)获批上市,自此改变了FDA审批Follow-on CDx的策略。与此前HER2检测常用的一致性研究策略不同,F1CDx是通过与原研CDx——cobas EGFR Mutation Test对比做非劣效研究,证明其性能不明显劣于原研CDx,从而作为Follow-on获批。非劣效研究在评估申报方法和参考方法之间的差异时,也会将参考方法本身的波动考虑在内,二者之间的差异只要符合预先设定的非劣效边界要求,即可获批。F1CDx之后的很多CDx Follow-on申报,都被要求按照该研究范式进行。
对于HER2检测的Follow-on申报来说,过去常遇到的问题是:原研CDx阅片人判读结果之间方差较大、一致性研究通过难度较高。至此,相当于FDA提供了两种Follow-on获批方法供HER2检测选择——与原研CDx对比做一致性研究,或非劣效研究。
04“药以群分”——Class labeling
部分FDA批准的非小细胞肺癌EGFR 19del或L858R CDx及相应药物
随着获批的靶向EGFR药物及其CDx数量增加,新的问题日渐凸显:医生需要先确定用哪种药,才知道应该做哪个CDx检测。由此推动了Class labeling(“药以群分”)的政策落地。2020年FDA发布了针对Class labeling的指南。其核心思路是,如果某CDx在同一个位点上已经有两个或两个以上的药物获批,即已有足够证据证明该CDx可以准确地检测此位点变异,则后续同适应症、同靶点、与靶点互作关系相同的药物可以直接加到该CDx的使用适应症中。FDA网站会更新相应清单。
CDx监管政策变化展望
以上,我们通过摘取一些既往的经典审批案例,对FDA CDx监管政策及审批策略做了简单介绍。不难看出,随着技术与应用场景不断变迁,FDA针对CDx的监管政策也在发展变化。那么未来还会有哪些改变?
2022年11月,FDA肿瘤学卓越中心(OCE)的Richard Pazdur博士公开表示FDA正在进行一个新的试点项目,提出最低性能标准(minimum performance criteria)概念,期望进一步打破癌症治疗中“一药一伴随”的固有方式。
需要注意的是,这个试点项目意味着未来做伴随诊断的方式可能会发生变化,并不是说未来不再需要做伴随诊断。未来具体怎么做,可能还存在不确定性,这与美国LDT的监管与使用现状有关。过去FDA曾通过510(k)为一些产品(如MSK-IMPACT、PGDx elio tissue complete等)提供了上市许可,但是这些产品并不能作为CDx指导用药。FDA希望这类性能已受认可的产品或服务可以用于临床患者的样本检测,积累真实世界数据,为未来的CDx用途审批或药物审批提供有效依据。
然而遗憾的是,目前美国临床实际使用的检测中,很多是性能未经FDA评审的LDT检测。关于产生这种监管现状的原因、FDA在此过程中作出的努力以及未来美国LDT监管趋势的讨论,我们将在下一篇推文——美国LDT和CDx监管现状与未来继续分享。值此国内LDT政策逐步落地完善之际,希望下一篇讨论能够为行业内人士提供一些参考和启发。
参考文献(滑动)
[1]Papadopoulos, N., Kinzler, K. W., & Vogelstein, B. (2006). The role of companion diagnostics in the development and use of mutation-targeted cancer therapies. Nature biotechnology, 24(8), 985–995. https://doi.org/10.1038/nbt1234
[2]https://www.federalregister.gov/documents/2011/07/14/2011-17671/draft-guidance-for-industry-and-fda-staff-on-in-vitro-companion-diagnostic-devices-availability
[3]https://www.fda.gov/regulatory-information/search-fda-guidance-documents/in-vitro-companion-diagnostic-devices
[4]https://www.fda.gov/regulatory-information/search-fda-guidance-documents/principles-codevelopment-in-vitro-companion-diagnostic-device-therapeutic-product
[5]Li, M. (2015). Statistical consideration and challenges in bridging study of personalized medicine. Journal of Biopharmaceutical Statistics, 25(3), 397-407.



