






医疗自动化制造设备展浅谈医疗器械生物相容性评价
2023-08-18
生物相容性是指医疗器械或材料在一个特定应用中引起恰当宿主反应的能力。“生物相容”与“生物不相容”不是某种材料天然的或绝对的“标签”,而是需要结合材料的具体性能和特定的(临床)应用场景进行判断
医疗器械设计开发过程中对于终产品进行生物相容性评价是必不可少的的环节,因其周期往往较长,评价的充分性和完整性往往是影响整个设计开发过程至关重要的环节。基于此,作者针对性的对于生物学评价常用的生物学试验谈谈在实践过程中的一些心得,避免大家踩同样的坑。

首先我们结合器审中心中涉及生物学评价方面的问题,先针对性的看看大家关注的问题以及器审中心的回答。我们在实际过程中最想省掉的可能就是生物学试验了,那么我们先看下哪些材料可以豁免生物学试验呢?
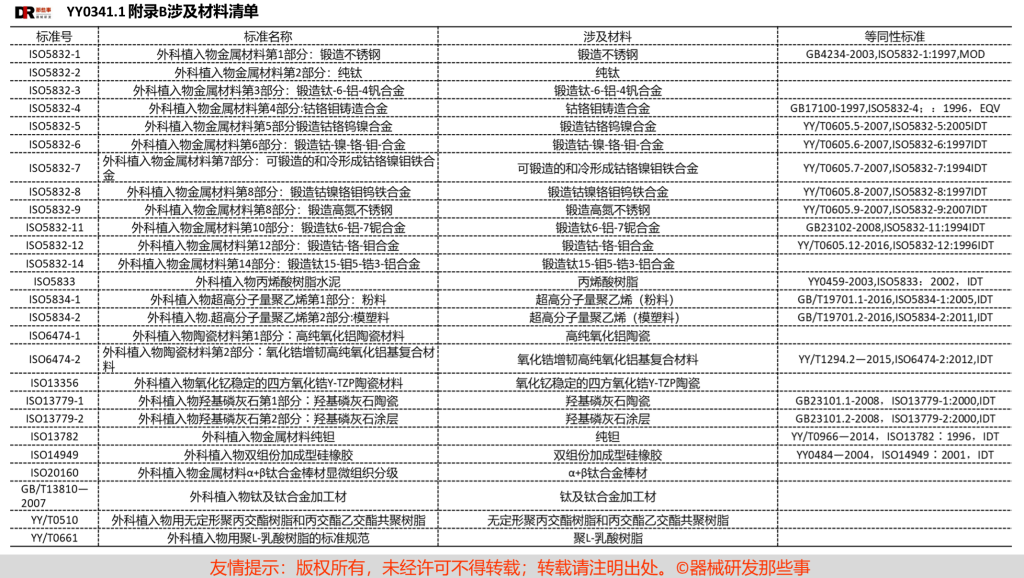
生物学评价不能豁免,可通过等同性比较,证明申报产品与已上市产品具有相同的生物相容性,从而确定申报产品的生物学试验的减化或免除。对于符合YY 0341.1附录B的材料,仍需通过等同性比较,如论证生产过程是否引入新的生物学风险,两者的生产过程(加工过程、灭菌过程、包装等)是否相同,因为生产过程也可能会引入新的有害物质(灭菌剂、加工助剂、脱模剂等残留物),若经评价,生产过程不引入新的生物学风险,则可认为豁免生物学试验。
看到这里是不是觉得很爽,其实并不是所有材料都需要进行生物学试验的,是有许多材料可以豁免生物学试验的。
全球高值医用耗材市场规模成长迅速,医用材料及部件受下游应用市场需求增长,或将迎来市场元年,医疗自动化制造设备展特设材料,部件和加工设备专区,品类范围包括原材料、成型材料、转接器、刀片、夹紧用品、管接头和连接件、紧固件、垫片/圈、量规,测量仪表,计数器、铰链和插销、嵌件、封堵物、光学部件、移液器、密封件等,目前已有路博润、塞拉尼斯、迈图、奥美凯、Acme Monaco、Pulse、Wytech、韦恩堡、田中贵、罗信、康德莱等代表性企业实力加盟。点击立即加入医疗自动化制造设备展。
此外,我们也可以选择已上市同等产品(通常为自己家的已上市产品,别家的通常也很难拿到等同性证据)的数据,但我们需要考虑一些影响生物相容性风险的因素,具体可参考器审中心的答复。
(1) 影响生物相容性风险的主要因素有:产品的材料化学组成(包括各组成材料比例)、产品物理结构、表面特性、生产工艺、灭菌方法、原材料供应商及技术规范、液体类产品/湿态保存类产品还需考虑内包装材料。
(2)若受试品与申报产品在以上所列可能影响生物相容性风险的因素中存在不一致的情况,则需提供充分的理由和证据支持所提交的试验报告适用于申报产品,必要时补充相应的生物学评价资料,如可沥滤物分析及毒理学风险评定资料、相关生物学试验项目的补充试验等。
根据医疗器械分类(人体接触类型及接触时间)选择对应的生物学评价终点(即生物学试验项目),如下表所示。
![]()
值得一提的是,关于慢性毒性,其周期是生物学试验中周期比较长的试验项目,同时也容易存在一定的失败性,可采用化学表征+毒理学评价的方式代替,这一点在器审中心发表的论文(医疗器械生物相容性评价:现状、进展和趋势,中国医疗器械信息,2021,27,11)中有提及,感兴趣的可自行下载查看。此外,对于生殖和发育毒性以及降解是需要根据材料的特性进行,而不是都不需要做。比如对于新材料或材料有已知的生殖或发育毒性,或者器械用于孕妇,则需要进行生殖与发育毒性测试;若材料存在降解可能的,就要提供降解信息。 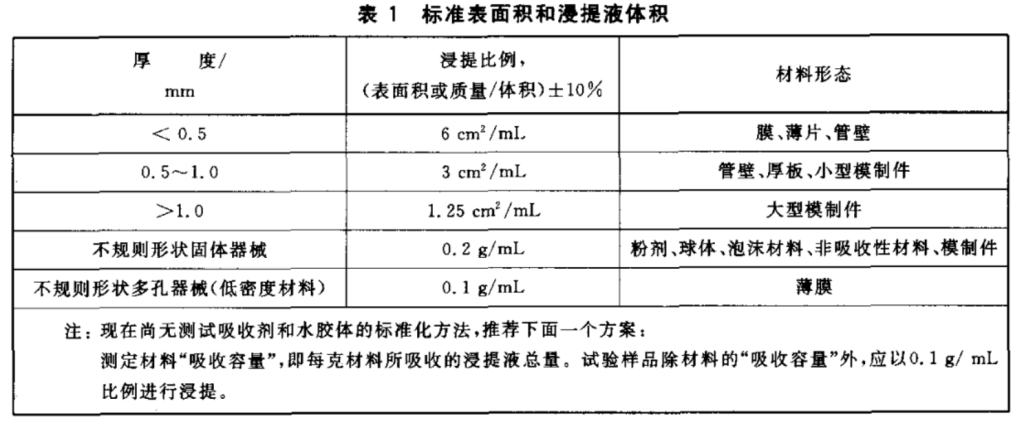
同时,对于不同类型或与组织持续接触时间不同的器械部件,需要分别浸提和测试。这一点非常常见也非常重要,比如某一产品存在输送和植入部分,则输送和植入部分需要分别进行测试,也就是需要根据GB/T 16886.1附录A的表进行生物学评价终点分别选择和测试。对于长期(大于24h小于30天)或持久接触(大于30天)的医疗器械,建议采用72h浸提时间,同时对于植入部分,浸提温度建议选择55±2摄氏度。同时,我们最关心的浸提浓度,通常我们会被告知为0.2g/mL(来自GB/T 16886.12表1,可能因为我们的器械为不规则形状),但关于浸提是使用表面积还是重量,建议优先选择表面积(至少FDA也是认可的,如果申报FDA建议选择表面积),具体的可参考ISO 10993-12。此外,我们也看下器审中心关于浓度的答复。
对于试验中出现统计学差异的评价指标,试验报告需明确相关差异是否有生物学意义并提供理由、分析判断相关差异与受试产品的关系,而非仅简单列出具有统计学差异的项目。另外,对于通过植入方式接触受试品的亚慢性毒性试验,需提供植入剂量的确定依据,如,在动物可耐受情况下,推荐样本植入剂量为拟用人体临床剂量的50~100倍。
浸提溶剂的选择也是生物学试验中至关重要的,对于浸提溶剂CMDE有相应的答复,值得注意的是这是2018年的答复。GB/T 16886.3已更新至2019版,如果没有记错的话也是需要双浸提的。
对于细胞毒性试验,由于含血清培养基是支持试验体系中细胞生长的必需介质,且具有浸提极性和非极性两种物质的能力,应当考虑作为细胞毒性试验首选浸提介质,此种情况下可仅选用含血清培养基一种浸提介质。对于致敏试验、刺激或皮内反应试验、急性全身毒性试验等项目,需考虑选择极性、非极性两种浸提介质;对于遗传毒性试验,根据GB/T 16886.3标准规定,适当时,应使用两种适宜的浸提溶剂,一种是极性溶剂,另一种是非极性溶剂或适合于医疗器械性质和使用的液体,两种溶剂均应与试验系统相容。
此外,我们在设计开发过程中常见的问题,比如是否可以采用原材料或替代性样品进行生物学评价,这个我们可以看看器审中心的答复。值得一提的是因为植入样品常常采用替代样品进行,尺寸要求在标准中有明确的规定(具体参照GB/T 16886.6),因此也常常成为发补的重点。因此,我们在生产植入样品时,建议拟定一个制备方案,并在其中从植入试验剂量、样品表面特性等方面综合分析植入试验能否代表种产品的局部植入反应风险。
生物学评价应考虑产品制造所用材料、预期的添加剂、工艺污染物和残留物、可滤沥物质、 降解产物、最终产品的物理特性、各个组件及他们在最终产品中的相互作用、包装材料和保存介质对生物相容性的影响等因素,因此产品的生物相容性试验原则上应采用终产品进行或采用取自最终产品上有代表性的样品。如采用终产品进行试验不可行,可考虑采用与终产品以相同的工艺过程制得的试样进行试验,但需对试样的代表性进行充分的分析论证。另外,当一个器械上有不同的组成材料时,在选择试验样品时应 考虑不同成分间可能存在的化学反应,以及不同成分对人体的综合作用。但若医疗器械不同组件与人体接触性质和接触时间不同,应考虑分别进行生物学试验。
至此,我们讲完了生物学试验,那么一份完整的生物学评价报告应包含哪些部分呢。这一点我们可以从医疗器械注册申报资料要求及说明中找到答案:
(1)描述产品所用材料及与人体接触性质,设计和生产过程中可能引入的污染物和残留物,设计和生产过程中可能产生的析出物(包括滤沥物和/或蒸发物)、降解产物、加工残留物,与医疗器械直接接触的包装材料等相关信息。
(2)描述申报产品的物理和/或化学信息并考虑材料表征(如适用),如器械的物理作用可能产生生物学风险,应当进行评价。
(3)生物学评价的策略、依据和方法。
(4)已有数据和结果的评价。
(5)选择或豁免生物学试验的理由和论证。
(6)完成生物学评价所需的其他数据。
医疗自动化制造设备展强势会议材料论坛将结合生物医用材料、精密配件及生产中精密的加工工艺在医疗器械中的应用为器械生产商和供应商提供交流平台。到此通过生物学试验进行的生物学评价就介绍完毕了,那么待后期产品上市后,如在实际生产过程中,修改了原材料或工艺发生了变化,是否需要重新进行生物学评价呢?我想答案是肯定的,基本原则就是当我们的风险产生了变化则需要重新进行生物学评价,这一点与风险管理一样是贯穿产品整个生命周期的。具体需要评价哪些问题可参考《无源医疗器械产品原材料变化评价指南》(2020年第33号)。
文章来源:器械研发那些事



