






国际医疗器械展览会|第二类有源医疗器械注册全流程审评解析
2025-05-16
近年来,第二类有源医疗器械的注册申报通过率持续承压,在首次注册、变更注册及延续注册申请中,不少产品因资料不全或不合规被要求补正。因此,国际医疗器械展览会结合审评案例与法规要求,深度解析三大注册类型(首次、变更、延续)的高频问题、典型案例及应对策略,助力企业高效完成申报。

图片来源:研检方程式
一、首次注册:技术资料“全面性”是核心,三大高频问题解析
首次注册是企业产品进入市场的“入场券”,但技术资料的完整性和规范性往往成为“拦路虎”。以下是审评中最常见的三类问题:
1. 综述资料:逻辑不清,关键信息缺失
典型问题:
产品作用机理描述模糊:例如某“医用红外热成像仪”未说明红外传感器与图像处理算法的联动逻辑,仅笼统描述“通过温度成像辅助诊断”。
结构组成不完整:某“无线生理参数监测仪”未明确软件架构(如嵌入式系统版本、数据传输协议),导致无法评估网络安全风险。
避坑策略:
技术原理图:标注核心部件(如传感器、处理器)的功能链路。
软件架构图:明确前端界面、后台算法、数据存储的交互逻辑。
按模块化编写:参考《医疗器械注册申报资料要求》,将综述资料拆分为“产品概述”“技术原理”“结构组成”“适用范围”四大模块,每模块需包含:
模板化对比表:提供与《免临床目录》产品或已上市同品种的对比表,量化差异项(如精度、响应时间)。
2. 研究资料:验证不充分,风险分析流于形式
典型案例:
某“高频电刀”性能研究仅测试基础切割功能,未评估不同组织模式下热损伤范围,导致临床使用风险未被识别。
某“睡眠呼吸监测仪”软件未提交漏洞扫描报告,后因数据传输加密等级不足被发补。
避坑策略:
硬件:新增材料(如钛合金电极)需提供理化性能(导电性、耐腐蚀性)及生物相容性(细胞毒性、致敏性)双验证。
软件:依据《医疗器械软件注册审查指导原则》,提交以下资料:
核心算法白盒测试报告(如心电图QRS波检测算法的灵敏度/特异性验证)。
网络安全渗透测试结果(如模拟攻击下数据篡改风险)。
差异化验证设计:
风险分析模板:采用FMEA(失效模式与影响分析)框架,列出所有潜在风险点(示例):
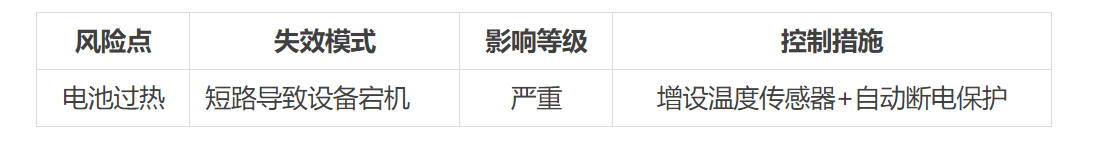
3. 临床评价:路径选择错误,对比证据不足
典型案例:
某“动态血糖监测系统”误将产品归类为《免临床目录》中的“血糖试纸”,因监测原理(微创 vs. 无创)差异被驳回。
某“超声骨密度仪”同品种对比仅描述“精度相近”,未提供统计学差异分析(如Bland-Altman图)。
避坑策略:
三步法确认路径:
查目录:严格对照《免临床目录》中的“产品描述”“适用范围”“技术特征”三项,任何一项不匹配即需临床数据。
选同品种:优先选择境内已上市、注册证信息完整的产品作为对比对象。
量化差异:使用表格明确申报产品与对比产品的差异项,并提供支持性数据(示例):
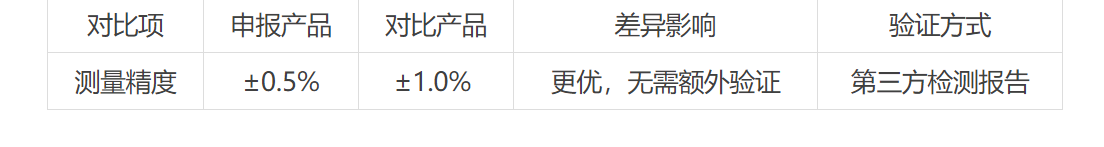
二、变更注册:细节决定成败,三类“致命”疏漏
变更注册是企业产品迭代的关键环节,但注册单元划分、检验覆盖不全等问题频发:
1. 注册单元划分错误:技术原理差异被忽视
典型案例:
某“医用电子内窥镜”新增4K成像功能,因图像处理芯片从FPGA升级为ASIC,被判定为不同注册单元,需重新注册。
某“呼吸机”新增高流量氧疗模式,因工作原理(涡轮驱动 vs. 压电驱动)变化,未通过变更注册。
避坑策略:
四要素对比法:从“技术原理”“结构组成”“性能指标”“适用范围”四个维度,提交变更前后对比表。若任一维度存在实质性差异,需拆分注册单元。
预判管理类别:例如软件新增AI辅助诊断功能,需评估是否符合《人工智能医疗器械审评指导原则》中的Ⅲ类要求。
2. 检验报告:覆盖不全,方法过时
典型问题:
某“多参数监护仪”新增血氧饱和度检测模块,仅检测单一型号,未覆盖全部规格(成人/儿童探头)。
某“激光治疗仪”波长调整后,仍采用旧版GB 7247.1-2012标准检测,未同步更新至2024版。
避坑策略:
三阶检测法:
全项检测:针对变更部分(如新增模块)执行全性能测试。
差异检测:对未变更部分,抽取典型型号验证一致性。
方法更新:若标准换版,需提交新旧方法等效性报告(如检测精度偏差≤5%)。
3. 临床评价:新增功能“想当然”
典型案例:
某“心脏起搏器”新增远程监护功能,企业认为属于“软件升级”未提交临床数据,后因缺乏电磁兼容性对人体影响的验证被发补。
某“输液泵”流量精度从±5%提升至±2%,未提供临床获益证据(如减少输液反应发生率)。
避坑策略:
功能影响分级:
A类(高风险):涉及诊疗决策(如自动给药剂量调整)→需临床试验。
B类(中风险):性能优化(如精度提升)→需同品种对比+文献支持。
C类(低风险):界面优化(如语言切换)→无需额外数据。
三、延续注册:警惕“标准更新”与“资料一致性”
延续注册看似简单,但标准更新和资料疏漏常导致发补:
1. 强制性标准未同步更新
典型案例:
某“超声洁牙机”未将YY 0460-2023《牙科设备安全要求》纳入技术要求,因振动频率超标被要求补检。
某“电动手术床”引用已废止的GB 9706.15-2008,未更新至GB 9706.1-2020通用标准。
避坑策略:
标准动态监控表:建立企业标准库,定期核查有效性(推荐工具:国家标准全文公开系统、NMPA标准管理中心)。
过渡期应对:若新标准实施后旧标准仍有效,需提交声明明确执行版本及理由。
2. 申请表与附件信息矛盾
典型问题:
某“血糖仪”延续注册申请表中生产地址为“A市”,但附件厂房产证显示“B市”,因信息不一致被发补。
某“电子血压计”分类编码误填“07-03-01”(无源器械),实际应为“07-05-02”(有源器械)。
避坑策略:
四步自查法:
一致性核对:申请表、说明书、技术要求中“产品名称”“型号规格”“结构组成”需完全一致。
分类编码确认:通过《医疗器械分类目录》查询工具(如NMPA官网)复核编码。
证照更新:如生产许可证变更,需同步提交最新版本。
电子签名校验:PDF文件需确保所有签章清晰可辨,避免扫描模糊。
3. 符合性声明不完整
典型案例:
某“雾化器”声明中仅提及“产品无变化”,但实际包装材料从PVC更换为TPU,未说明“变更通过质量管理体系控制”。
某“康复机器人”未声明符合最新行业标准《YY 9706.278-2023 医用电气设备 第2-78部分:康复、评定、代偿或缓解用医用机器人的基本安全和基本性能专用要求》。
避坑策略:
五要素声明模板:
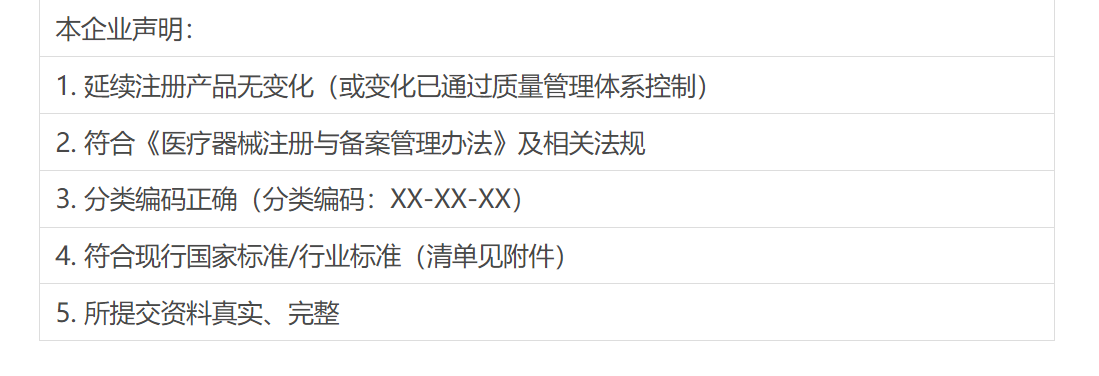
四、企业合规管理:从“救火”到“防火”的三大升级
1.建立注册全生命周期档案:
使用数字化工具(如PLM系统)管理技术文档,实现版本控制、变更追溯。
定期更新法规库,设置标准更新自动提醒功能。
2.内部预审机制:
组建跨部门审评小组(研发、质量、注册),模拟发补场景,提前修复漏洞。
参考《医疗器械注册自查表》,逐项核对资料完整性(示例自查项):
综述资料是否包含结构示意图?
研究资料是否覆盖所有性能指标?
检验报告是否签字盖章?
3.外部专家支持:
与第三方法规服务机构合作,获取最新审评趋势解读(如AI医疗器械、无线通信设备的特殊要求)。
参与行业协会培训,学习典型案例(如某企业因未提交网络安全事件应急预案被发补)。
结语:细节控+体系化=高效过审
第二类有源医疗器械的注册不仅是合规要求,更是企业技术实力与管理水平的体现。从首次注册的“全面性”、变更注册的“精细度”,到延续注册的“持续合规”,每个环节都需“细节控”思维。建议企业摒弃“应付式”申报,转向“体系化”管理,结合内部预审与外部赋能,最大化降低发补风险,加速产品上市进程。
文章来源:研检方程式
若涉及侵权,请立刻联系删除


