






2023上海医疗器械创新展全面解析医疗器械进口美国的FDA认证要求
2023-09-14
医疗器械FDA认证对于产品进口到美国的要求是非常严格的。作为美国食品药品监督管理局(FDA)对医疗器械质量和安全性的认可,FDA认证是医疗器械在美国市场合法进口和销售的重要条件。
医疗器械范围很广,小到医用手套,大至心脏起搏器,均在FDA监督之下。可能大家都了解美国进口医疗器械需要FDA认证,但是大家对于美国医疗器械的分类可能会存在一些疑问:为什么我的产品都没体现医用也要进行FDA注册?今天我们就来细说一下关于美国医疗器械分类,并进一步阐述医疗器械进口美国的清关要求以及FDA注册流程!

医疗器械的分类
医疗器械是指符合以下条件的仪器、装置、工具、机器、设备、植入物、体外试剂或其他相关物品,包括组件、零件或附件:明确列于National Formulary或the Unite States Pharmacopeia或前两者的附录中;预期使用于诊断、治疗、预防、治愈或缓解动物或人类疾病;预期影响动物或人体身体功能或结构,但不经由新陈代谢来达到其主要目的。
|
|
|
|
|
Ⅰ类器械 |
牙线、刮舌器、牙刷、冲牙器、洗鼻器、拐杖、吸乳器、眼镜框、眼镜片、太阳眼镜、弹性绷带、压舌板、听诊器、雾化器、检查手套等 |
一般控制 |
|
Ⅱ类器械 |
脱毛仪、医用外科口罩、手术服、注射器、血氧仪、助听器、体温计、血压计、心电图仪和输血输液器具等 |
一般控制和特殊控制 |
|
Ⅲ类器械 |
人工耳蜗、自动体外除颤器(AED)、人工晶体、心脏起搏器和植入式输液泵等 |
一般控制和上市前批准 |
医疗器械的清关要求
FDA认证是一个统称,根据产品的类别不同,FDA认证要求也会不同,比如食品类产品的FDA认证要求是需要制造商进行FDA注册以及进口商需有邓白氏编号等。
● 制造商必须进行FDA注册,并列名产品
● 初始进口商也需进行FDA注册
● 根据医疗器械的类别不同,还需510(K)上市前通知或PMA上市前批准
● 在进口清关前,已注册的初始进口商需通过制造商的企业注册号或企业名称和地址、产品注册号与制造商及产品进行关联。
不能出现有虚假或误导性的标签,除了在波多黎各或主要语言不是英语的美国领土内分销的产品外,所有标签应该通过英文呈现。
标签必须包含以下内容:
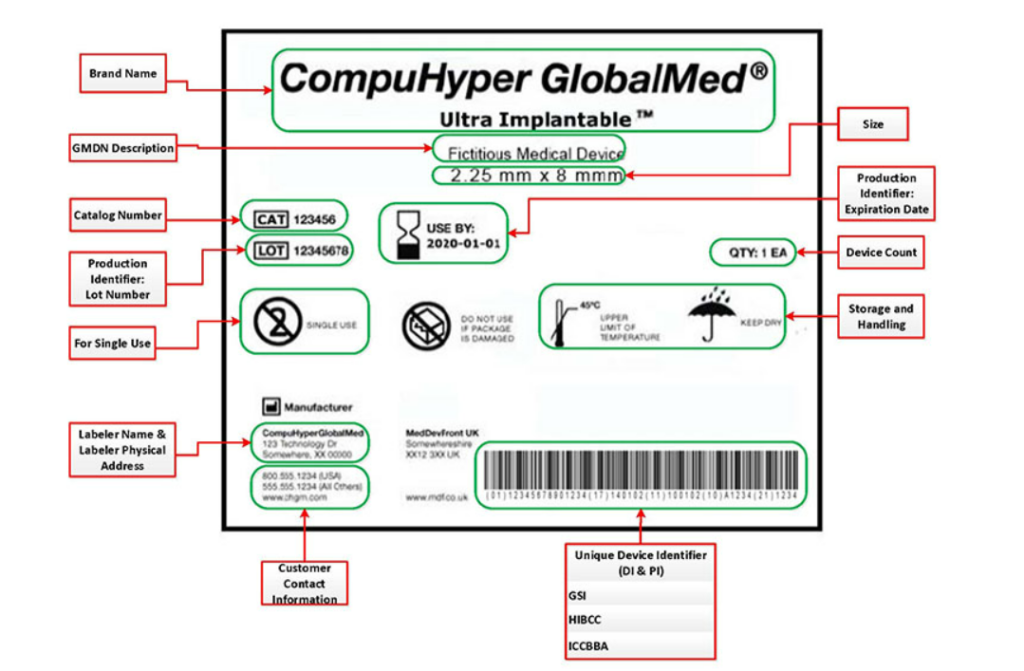
2023年医疗器械注册和监管会明确了重点的工作方向,法规建设、医疗器械创新审批、上市前和上市后监管依旧是国内法规的重点。为保障医疗器械企业的安全监管,2023上海医疗器械创新展Medtec创新展设立 “中外医疗器械政策法规” 、“中外医疗器械质量”等论坛,保证企业创新产品的合规上市,助力医疗器械全生命周期各环节质量安全监管。点击立即报名参观2023上海医疗器械创新展Medtec创新展。
医疗器械FDA注册

数据库链接:
https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfPCD/classification.cfm
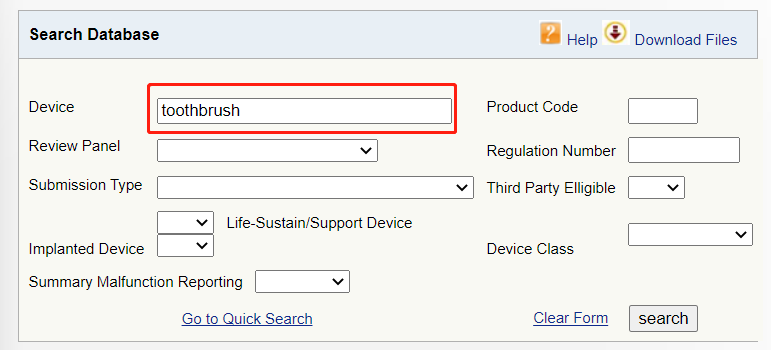
▶ 在列表中选择合适的设备,如果是电动牙刷,点击“toothbrush,powered”超链接进入
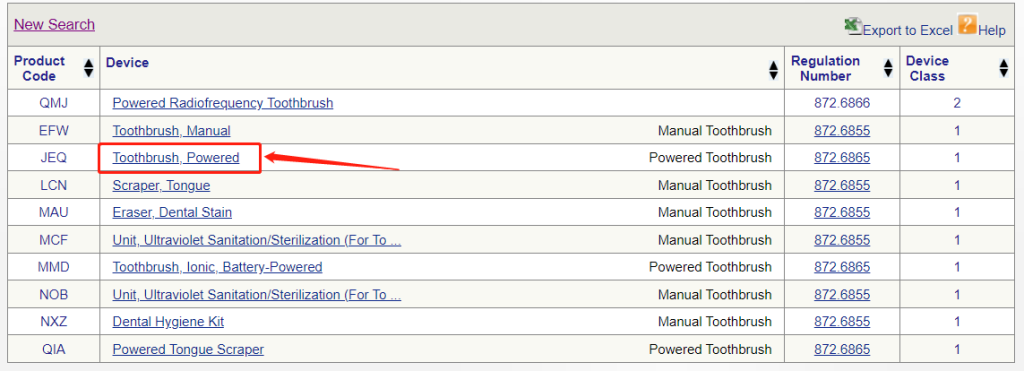
▶ 查看产品分类信息,电动牙刷属于Ⅰ类医疗器械,且豁免于510(k) 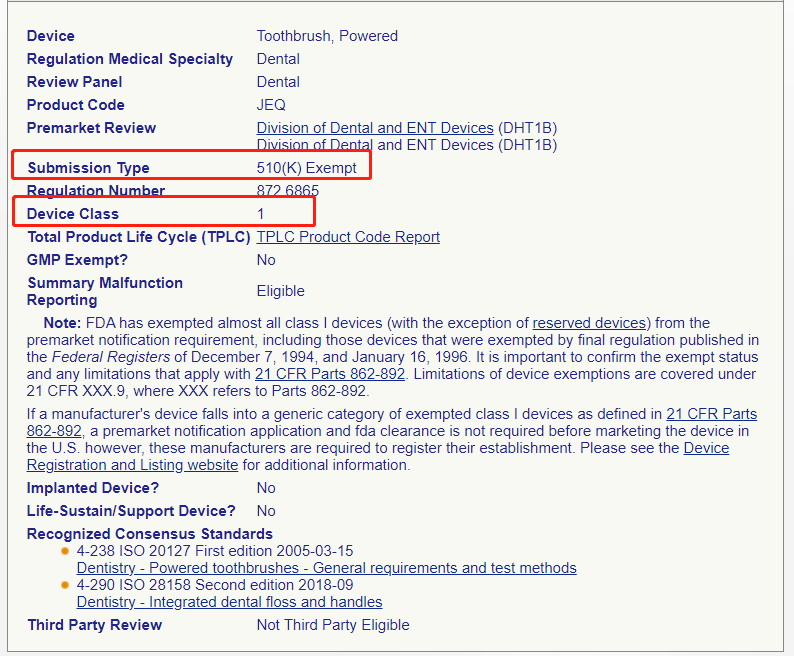
Ⅰ/Ⅱ类豁免产品直接进行工厂注册和产品列名,某些 Ⅰ类和大多数 Ⅱ类设备需要准备 510(k)资料,Ⅲ类设备需要准备PMA资料。
被要求提交510(k)和PMA的Ⅱ类和Ⅲ类医疗器械,需要准备技术文件和性能测试报告等资料。Ⅲ类器械有可能需要进行临床研究,提交PMA后,FDA会对制造商和供应商进行现场考核。
完成注册后,FDA不会颁发FDA证书,但是会提供注册号给企业,当然有些第三方机构代为注册会提供一份证书给企业,但这份证书不是由FDA官网颁发的。无论是自己进行注册还是找第三方机构代理注册,都可以通过FDA注册号在FDA官网来进行查询。
官网链接:
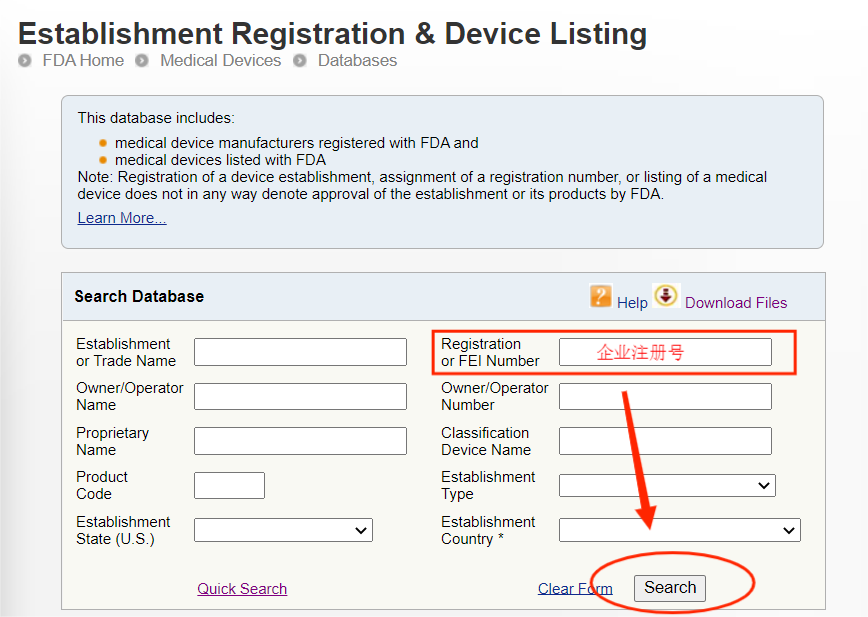
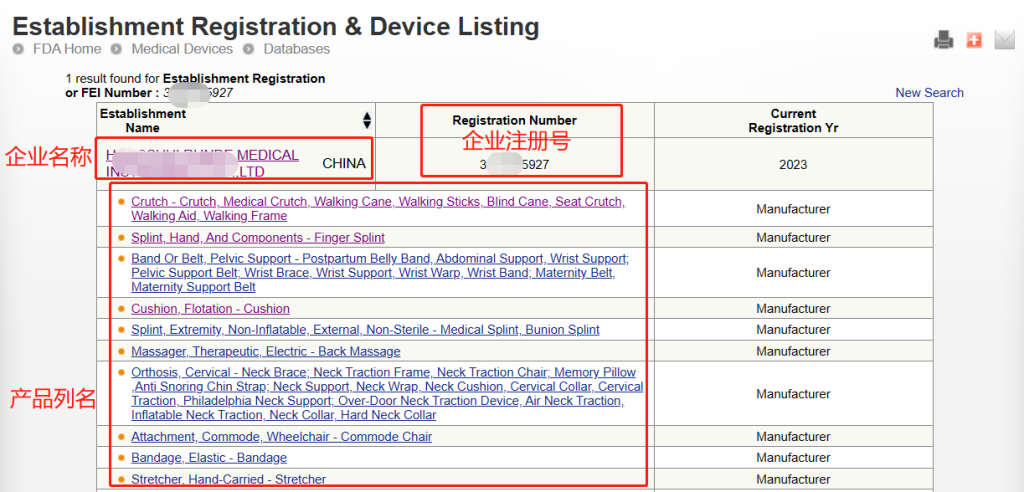
注意:此查询方式可查询到的是制造商、进口商的注册信息以及产品的列名,但产品注册号(Device Listing NO.)属于保密信息,是无法直接查询到的。
更多医疗器械法规以及监管条例解读,请关注2023上海医疗器械创新展Medtec创新展!
文章来源:九方通逊


