






中科院开发一种有机有源适应晶体管,可用于替换人类患者器官的设备
2021-08-10
日前,中国科学院北京分子科学国家实验室和中国科学院大学的一组研究人员开发了一种有机有源适应晶体管(OAAT),该成果已发表在《自然电子》杂志上。
论文中,该小组描述了他们如何克服涉及电荷传输的障碍,并探索了他们的 OAAT 的可能用途。
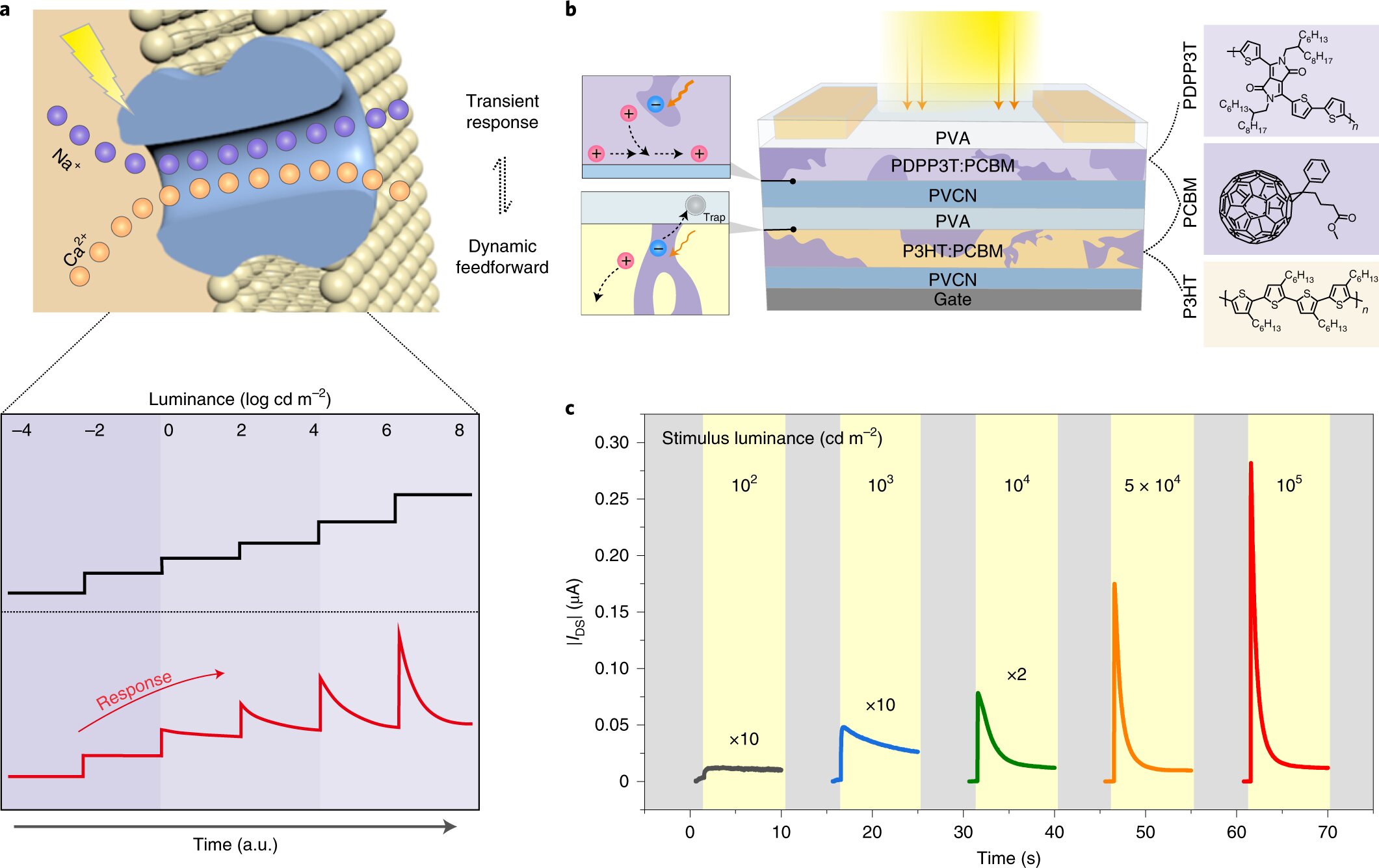
a,上图:人体光感受器的光响应示意图及响应特性。底部:单个亮度步骤中的个体响应。当亮度周期性地逐步增加时,响应遵循锯齿曲线。随着亮度的每次增加,反应会跳跃,然后恢复到眼睛适应亮度刺激时产生的平衡水平。b,具有两个互补 BHJ 的 OAAT 示意图。c,OAAT 对暗背景上各种刺激亮度的实时光响应。施加的 V DS和 V GS分别为 -1 和 -4 V。图片
来源:自然电子(2021)。DOI:10.1038/s41928-021-00615-8
人眼能够进行一种即时适应——例如,当从黑暗的剧院中移动时,眼睛会自动发生变化以对外面的明亮天气做出反应——这并不像改变画面那么简单。通过打开或收缩瞳孔让通过镜头的光量。眼睛后部也必须发生变化,在那里运输不同类型的离子。在这项新的工作中,研究人员试图通过创建一种照片自适应设备来复制这一过程——一种可能有朝一日用于恢复眼睛损伤患者视力的设备。
在开始他们的工作后不久,研究人员遇到了一个主要障碍——如何处理电荷传输的冲突需求——需要抑制和光激发。经过大量实验,他们提出了一个新想法——将两个体异质结作为器件的两个不同层引入。一个用作光响应有源层,另一个用作浮栅。
经过更多的实验,该小组想出了一个功能齐全的七层设备:第一层是聚乙烯醇 (PVA),一种电介质。在它下面是第一个异质结。接下来是聚(乙烯基肉桂酸酯)(PCVN),另一种电介质。然后是另一层 PVA,接着是第二个异质结,然后是另一个 PCVN层,最后是栅极。
一旦他们的设备完成,研究人员发现他们没有好的方法来测试它。经过三个月的讨论,他们提出了所谓的“主动适应指数”——它可以用来测试人眼的适应能力,然后与他们新开发的 OAAT 进行比较。他们发现这两个来源的分数非常相似。
研究人员表示,他们的工作代表了创建自适应设备的第一步,该设备可用于机器人技术和用于替换人类患者器官的设备。
来源:EDN电子技术设计


