






最新版ISO 10993-1生物学评价及化学表征16问
2020-08-24
1、ISO 10993-1和提交FDA审评文件区别?
美国FDA于2016年06月正式公布了生物学评估标准“ISO 10993-1标准的使用《医疗器械生物学评估—第1部分:风险管理过程中的评价与试验指南》Use of International Standard ISO 10993-1, "Biological evaluation of medical devices – Part 1: Evaluation and testing within a risk management process" 。
FDA升级的法规,对于部分测试评估及萃取要求和ISO 10993-1内容有差异,尤其是高风险可降解植入物产品需观察植入后样品降解的短、中、长期时间对于生物体的影响,和对于植入物或贴有无热源(non-pyrogen)产品的热原及内毒素试验质控有新要求。另外,FDA不承认ISO 17025体系,要求生物相容性测试必须在正规的GLP生物学实验室完成。因此要申请美国FDA注册的医疗器械生产厂家,务必要委托具有GLP资质的实验室,按照FDA生物学评价标准完成有关的生物学试验。FDA法规原文节录如下:
Any in vitro or in vivo biological safety experiments or tests should be conducted in accordance with recognized Good Laboratory Practice (GLP) regulations including, but not limited to, the assignment of competent trained staff in the conduct of biocompatibility testing.
2、材料的可沥滤物和降解产物有区别吗?
可滤出物指的是产品(材料、组件或器械)通过溶剂浸提后释放出来的物质;降解产物是由原始材料在生物体内化学裂解而产生的颗粒或者化学物质,可见两者定义是不相同的。
目前ISO 10993-1改版后最大的不同,需要在试验开展前考虑产品材料的全面信息资料,经过风险评估以及化学表征评价后(得到可沥滤物信息及毒理评估信息)再进行生物学测试。
3、牙科材料的Sub-chronic systemic toxicity试验,可以不做非极性浸提的吗?
目前ISO 10993-1法规要求生物学测试都需要用极性(polar)和非极性(non-polar)两种溶剂浸提及实验,所以牙科材料的亚慢毒性也需要评估两种浸提液的毒性反应。在ISO 7405牙科系列标准有提到亚慢毒建议用经口服方式投予,若为牙科产品注册建议参考此标准。
4、重复使用产品,每次使用不指定同一患者,接触累积时间和用量如何考虑?
ISO 10993-1改版后有特别提到,重复使用的医疗器械要依照产品接触病人的累积时间评估风险,此要求会针对单一患者接触的时间计算。若不指定同一患者,可根据临床上实际使用的情形进行生物学风险评估。
5、非无菌提供的产品,在做细胞毒试验之前为什么要灭菌?
细胞毒性试验常常会因微生物的干扰而造成假阳性的结果。因此,准备样品时一定要保证环境的清洁及产品包装的完整性,才能降低细胞毒试验失败的风险。若产品上市后是非无菌的医疗器械,建议模拟产品实际上市状态,送测前先做简单的清洁后再进行测试。若担心产品有微生物需要灭菌,则需要评估灭菌对于材料所产生的影响。
6、毒理学评价具体的流程?是否可以举例?是否可以采用风险管理中的生物学风险的评价来进行?
ISO 10993-1:2018版发布后,进行材料化学表征分析为生物学评估的第一步,并需要对可沥滤物的可允许限量进行推导和毒理学评估以及风险控制。具体的流程依据图一进行(来源于ISO 10993-17图一)。毒理学评估建议由专业的毒理专家进行;风险管理的方式建议大家参照ISO 15499和ISO 14971的标准执行。
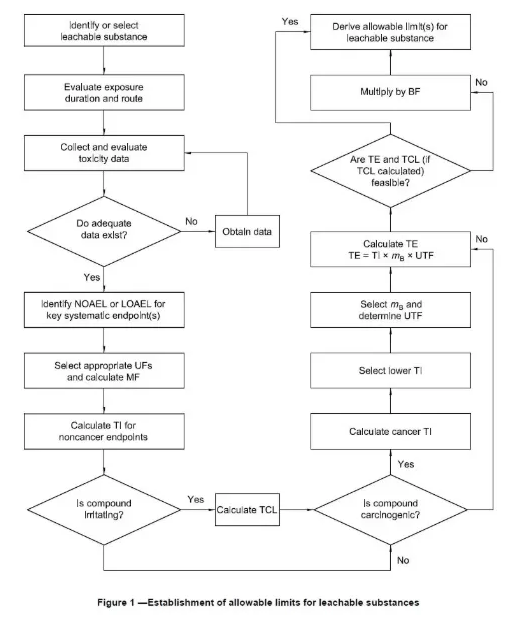
摘自ISO 10993-17 figure 1
7、如果条件不允许使用灭菌后产品进行试验,可以使用关键物料代替吗?
根据ISO 10993生物学标准要求,必须选用终产品或与终产品相同材料制成(包含灭菌方式)的样品进行测试;若仅采用关键物料测试,则需要给出合理的解释。
8、药物支架,测试细胞毒性,急毒时要不要含药测试,含药测试对急毒是否有影响?
含药器械需要先确认药品的安全性及与材料的药物相容性,然后根据法规以终产品的方式进行生物学试验。若担心药物对生物学试验的影响,则可以先收集药品相关安全性数据及文献后再进行测试。
9、创口贴类产品如果细胞毒性测试出来显示潜在细胞毒性,可以怎么进行下一步?
创口贴产品容易因胶体影响细胞毒性试验,建议可以收集相关的文献资料或是与已上市的等同样品的测试结果进行比较:若是轻度的细胞毒性(1分),则符合法规的要求(细胞存活率需在70%以上);若是毒性分数较高,则建议更换其他通过生物学测试验证的医疗级材料。
10、急毒,亚急,亚慢,慢性毒的区别,该如何根据产品进行选择?
这些系统毒性试验的选择差异基于评估产品接触病患的部位和时间。若是产品接触部位为组织粘膜或是体外连通器械、且接触时间小于24小时,仅需要做急毒试验;接触超过30天以上的医疗器械,除了要做急毒外还需要加做亚慢毒或慢性毒试验。
急性系统毒性仅观察产品短期的毒性反应,亚慢及慢毒主要是观察动物经连续投喂及处死后,用病理解剖方式确认组织器官是否有病变。若没有相关文献来佐证材料的安全性(如:新材料),则需要评估慢性毒性试验;反之,若有相关的安全性数据资料,则可以豁免。
11、生物学试验中试验动物的选择是否一定是需要采用双性别(器械本身不区分性别使用)?
生物学试验动物的选择,需根据ISO 10993测试标准的要求,有些测试需要进行单一性别(如:致敏性),有些测试需要采用双性别(如:亚急/亚慢毒性试验)。具体可根据产品的预期用途和接触时间,来评估对应的测试项目。
12、供应商提供了化学表征,器械生产厂家可以免于化学表征吗?如果供应商不能提供,是不是器械生产商必须要自己去测试?
若供应商提供了材料的相关化学表征资料,则可以引用到产品的风险评估报告中。但器械生产厂家还需要评估产品生产过程中的添加剂及残留物的风险。供应商若能提供相关资料,则可降低测试失败的风险。如果供应商无法提供相关报告,生产商则需要自行开展材料化学表征风险评估计划。
13、长期使用的产品超过30天“亚慢毒已经做了”,急性全身毒是不是一定要做,还是可以不做?
长期使用的产品根据10993-1评估表格,除了做亚慢毒外,还必须做急性全身毒性试验。急性全身毒性为观察产品接触生物体后的短期立即性毒性反应,与亚慢毒性评估的指标有差异。
14、15499与14971有什么区别?生物学评价需要引用15499吗?
ISO 14971和ISO 15499都有讲述风险管理的方式,14971讲的是医疗器材生命周期中风险管理的依据准则,15499则是针对生物相容性的风险评估流程,里面都有提到怎么用风险管理的手法去做生物相容性的评估。但风险是无所不在的,建议生产厂家结合两个标准,进行产品风险评估。
15、萃取条件的选择,温度的选择,比表面积的选择?
根据医疗器械的使用方式和产品型态,可依照ISO 10993-12: Sample preparation and reference materials要求,进行温度、表面积比例等萃取条件的选择。材料化学表征的萃取方式也可以参照此标准。
16、溶剂环己酮换厂家,这个应该怎么生物学评估?
若溶剂更换厂家,需要重新做风险评估分析(更换溶剂是会影响产品质量的重大变更),评估后若有需要生物学测试,确认产品安全性,则进行相应的实验,最后将测试结果汇总到生物学风险评估报告内。
来源:医用塑料


