






突发!迈瑞强势入局ECMO
2022-02-21
企查查APP显示,2月14日深圳汉诺医疗科技有限公司发生工商变更,新增迈瑞医疗关联公司深圳迈瑞股权投资基金股份有限公司为股东。
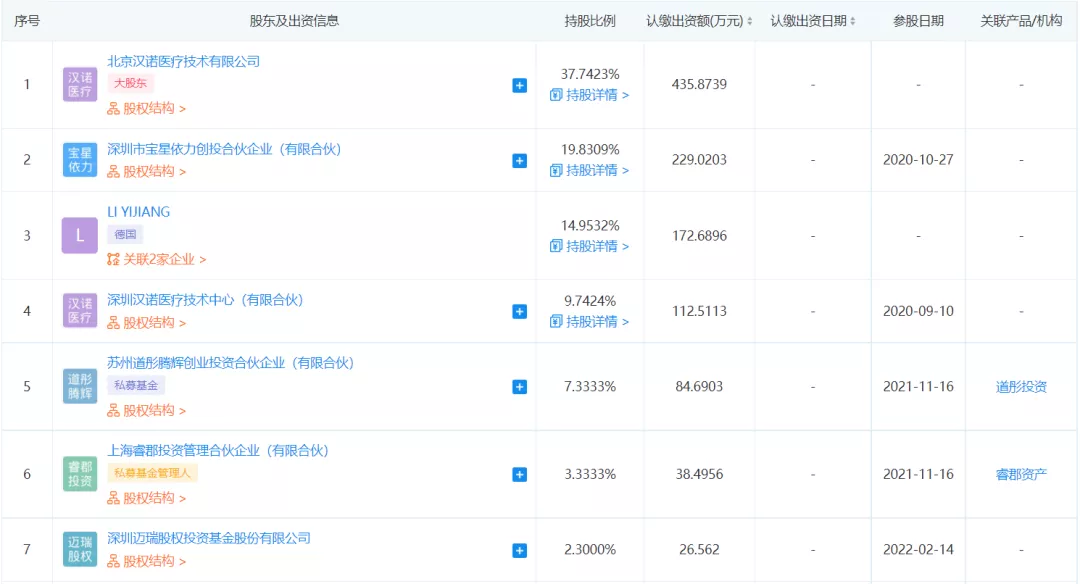
企查查信息显示,汉诺医疗是一家生物技术服务商,公司成立于2018年法定代表人为李鸣涛,注册资本1154.87万元,经营范围包含:二类、三类医疗器械的批发、零售;医疗器械的设计、开发、生产与销售等。

深圳汉诺医疗科技有限公司于2018年,由多位留德顶尖医疗技术专家创立, 重点从事针对 心脑血管 、器官支持和保护等重大方向的研发和制造,尤其是危机重症方面的高端医疗设备与耗材的创新研发和加工生产制造。致力于研发填补国产化空白的便携式生命急救系ECMO统,是国内少数研制整体ECMO系统的技术单位。

汉诺医疗刘洋董事长接受深圳卫视众创TV栏目采访
汉诺医疗的产品“Lifemotion 心动力”便携式体外循环系统项目在第八届中国(深圳)海归创业大会,得到9.47的高评分,荣获本次海归创业大会最具人气奖!
公司在 2020 年获批认定国家高新技术企业,近两年承担或参与了多项省、市、区级科研攻关专项,取得一系列科技创新成果。
据公司官网介绍,依托中德联合研发团队,经过3年科研攻关,已设计出氧合器和离心泵,目前完成了ECMO各核心部件包括氧合器、离心泵和系统主机在内的整套设备的功能样机的设计定型,即将进入技术转化和产品注册阶段。
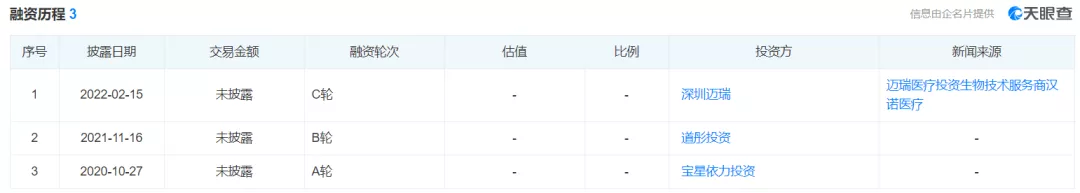
公司目前已完成了三轮融资
公司目前已经完成了三轮融资,其B轮融资投资方为道彤投资,道彤投资创始合伙人黄宁是医疗行业的老兵,曾担任武汉亚洲心脏病医院创始执行总经理(七年);曾创立上海国宾医疗控股有限公司,担任董事总经理,爱康国宾(Nasdaq:KANG)联合创始人。2017年,黄宁投资的凡迪生物被知名机构收购,投资三年获得近四十倍的回报。
道彤投资合伙人段涛,在妇产领域享有盛誉,曾任同济大学附属上海市第一妇婴保健院院长。
ECMO(体外膜肺氧合,俗称“人工肺”),新冠肺炎疫情来袭,ECMO因为屡屡出现在急救场景中而进入公众视线。
目前,这一重要的医疗设备我国全部依赖进口,其关键核心技术长期被国外垄断,设备及耗材价格昂贵,“ECMO一响,黄金万两”成为了很多人对ECMO的第一个认知,是卫生健康领域的“卡脖子”问题。
01
大企业下场可解垄断难题?
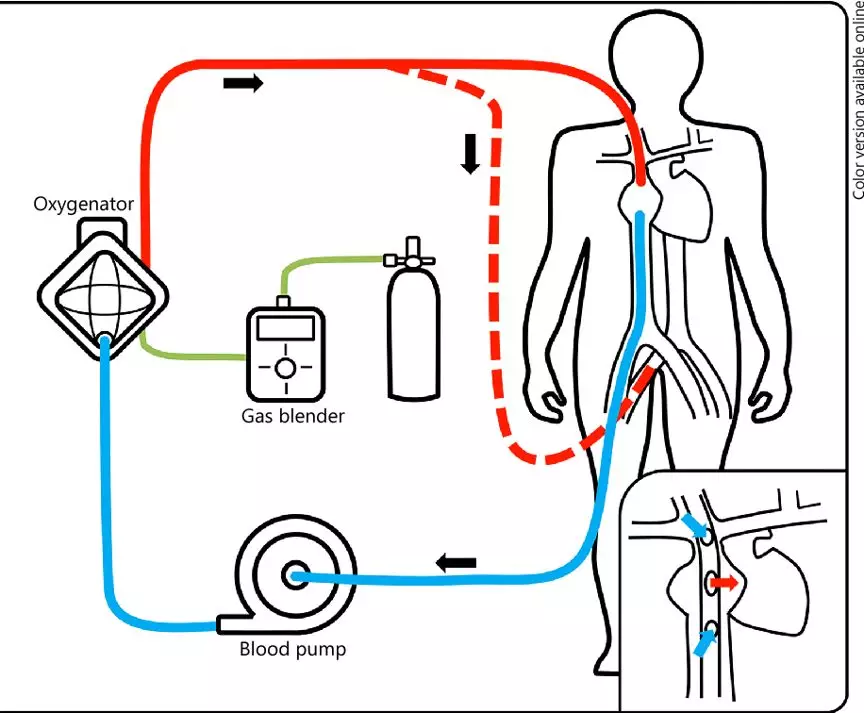
ECMO示意图
ECMO是心肺系统重症治疗的一种设备,原理是把静脉血引出体外,让血液与氧气充分氧合之后,再泵回体内,以代替心肺功能,可广泛应用于危重病人的急救。
武汉抗疫期间,ECMO(体外膜肺氧合,俗称“人工肺”)在医院临床使用成功救治重症新冠肺炎患者,并被纳入国家卫健委发布的新型冠状病毒肺炎诊疗方案,被誉为“救命神器”。

目前,国内市场主流的ECMO品牌主要是欧美的美敦力、米道斯、迈柯唯和索林等。根据中国医师协会体外生命支持专业委员会的数据,截至2018年底,国内共有ECMO设备400余台,其中迈柯唯和索林两家公司的产品占国内市场的绝大部分份额。
华西证券的报告显示,国内ECMO的均价为165万,而移动ECMO则需要约300万/台。这在医疗设备中并不昂贵,但ECMO的花费主要来自于使用过程中。
据业内人士介绍,ECMO开机耗材套包平均5万元,其中装有血液管路、膜式氧合器、离心泵头、插管及各类接头等,加上重症ICU每天超过1万元的治疗费,患者使用ECMO两周需要花费20万元。
使用价格高是制约ECMO广泛使用的重要原因之一。多名专家认为,设备国产化对降低使用费用和提升患者的接受程度至关重要。国产化能够提升市场竞争度,推动产品价格下降。

ECMO被认为是急救重症领域的最先进技术之一。其技术难点如下:
离心泵(人工心)
新一代离心泵的核心是磁悬浮技术和高效叶轮的设计,难点在于如何在提高泵血效率的同时降低泵头对血液的破坏和损伤,这需要精细复杂的生物医学工程技术支撑。
氧合器(人工肺)
而在氧合器的研发中,关键材料是聚甲基戊烯(PMP)的中空纤维膜,能够让氧气高效进入血液,代替人体肺部的气体交换功能。这些都有待科研攻关突破。
ECMO的研发需要时间和技术积累,作为一个相对小众的医疗器械,最终还是要看企业的战略、技术力量和经费。

美敦力ECMO系列产品
从全球排名前三的ECMO供应商看,美敦力全球第一,迈柯唯(Maquet)属于瑞典洁定(Getinge Group),索林(Sorin)之前属于英国理诺珐(LivaNova)——2017年底,由马云创立的云峰基金与微创医疗携手出资15亿港元将索林收购。两家公司都是长期专注心血管领域的企业。
第三方网站Medical Design & Outsourcing最新发布的《2021年医疗器械企业百强榜单》显示,全球营收最高的医疗器械公司还是集中在欧美地区。美敦力在2021年以301.17亿美元依然位居榜首,洁定收入32.38亿美元排名32位,理诺珐收入9.34亿美元位列全球69名。(点击查看详细排名)
今年由于财报公布时间问题,中国企业未有上榜,不过依迈瑞往年的业绩,全球排名预计在28到30之间。作为中国最大、全球医疗器械前 50 强企业,迈瑞有充足的实力支撑ECMO研发。
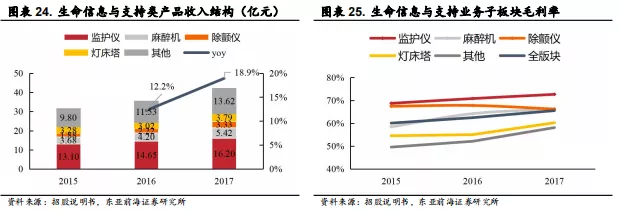
此外生命信息与支持产品是迈瑞三大业务中占比最高的基石业务,包括监护仪、除颤仪、麻醉机、呼吸机、心电图机、手术床、手术灯、吊塔吊桥、输注泵、手术室/重症监护室(OR/ICU)整体解决方案等用于生命信息监测与支持的一系列仪器和解决方案的组合。布局ECMO将进一步完善公司的产线,为患者提供更全面的支持。
02
企业踊跃响应号召
国产ECMO已完成首例临床
据业内人士介绍,ECMO是一个由不同部件组成的系统,一些企业能够生产其中的部件,而且这些部件都可以分开注册、单独使用。
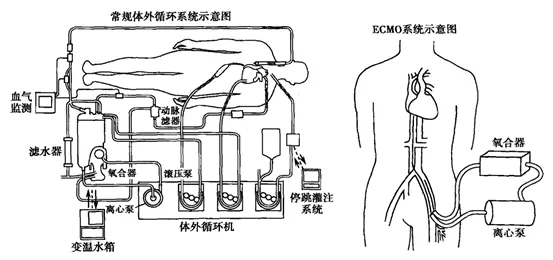
ECMO主要由膜式氧合器、离心泵组成,如果把它类比为铅笔和橡皮,中国企业已经分别会做铅笔和橡皮了,但还没有能力把它们结合起来。
2020年3月初,中国工信部召集了一次电话会议,有二十多家与ECMO相关的企业代表参会,交流各自的产品进展和研发进展,国家鼓励他们研发攻关,并表示会给予支持。
经过不到两年的努力,在近期国产ECMO已经传来了捷报!
2021年11月6日,西安交大一附院心血管病院应用中国首款自主研发的ECMO设备(体外膜肺氧合装置)成功救治两名重症心血管病患者。
2021年11月8日,医院公布,该院与四川大学国家生物医学材料工程技术研究中心、西安交通大学机械学院联合研发的体外膜肺氧合设备(ECMO)在国内率先进入临床阶段。
这款国产ECMO设备突破了国外的技术垄断,还运用了磁导航、磁控技术等新技术。西安交通大学机械学院庄健教授介绍,新冠肺炎疫情发生前,研发团队已基本完成ECMO离心泵设备及全系列耗材的样品试制,并进行了针对抗凝涂层的大动物实验。

2021年12月28日媒体获悉,航天长峰国家重点研发计划“ECMO系统研发”项目原理样机联调成功,获得内外部专家和临床医生的一致认可,标志着ECMO整体研发取得阶段性进展。
作为一家航天军工企业,航天长峰及其联合团队,先后完成体外膜肺氧合(ECMO)、热交换水箱、电子空氧混合仪、磁力耦合离心血泵、膜式氧合器等5种产品原理样机的国产化攻关研制任务,申请多项专利等知识产权。此次联调成功,是一次从材料、芯片电路到智能化集成等的全链条创新突破。
2018年,赛腾医疗来到苏州工业园区,开始了国产化ECMO研发。
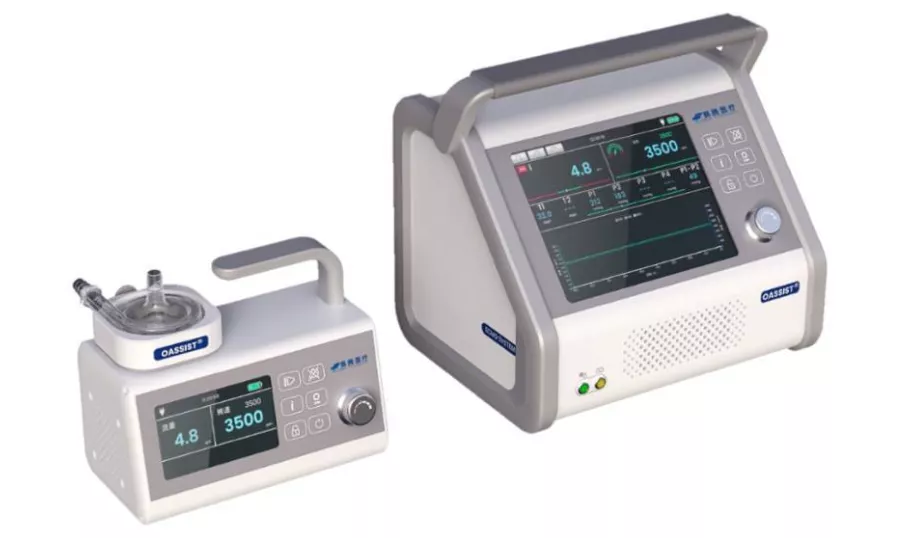
赛腾医疗OASSIST® 体外心肺支持辅助系统
2021年9月该公司生产的首款ECMO获得了中国医学科学院阜外医院伦理委员会批准,于2021年11月正式启动临床试验,今年1月初,一位24岁年轻女性患者出现心源性休克症状,考虑启动体外生命支持辅助流程。最终,国产的ECMO帮助患者心脏功能恢复,成功脱离体外膜氧合(ECMO)辅助。
目前,该系统正在进行多中心临床试验,预计今年就能完成注册并上市销售。而企业也已经建成厂房面积5200平方米,获批后即可投产,初步规划产能为1000套ECMO设备/年。
据参与了工信部电话会议的ECMO相关企业代表透露:目前各省的企业、高校已经行动起来,江苏、深圳已经有公司在申请了,大家热情很高,人人都在抢,最终要看谁能走在前面。
预计在未来,随着国家政策的驱动以及更多市场资本的进入,会激励更多的企业投入研发高端医疗器械,因此有望加快产品的研发周期。
03
患者数突飞猛进
近10年来,中国使用ECMO的患者数突飞猛进,病例数量从2004年的23例,跃至2016年的1234例。2018年发表在《中华医学杂志》上的文章显示:2017年中国(除台湾外)的233家医院开展了共计2826例ECMO,同比增加129%,开展的医疗机构数量增加了64%。而同年在国际ELSO(体外生命支持组织)注册的全球ECMO总例数则为9330。
中国生物医学工程学会体外循环分会2018年的统计数据显示,我国有260家医院可以做ECMO。但即使加上疫情后从国外紧急采购的ECMO,全国仅有500台左右,相当于平均每280万人拥有一台ECMO。
而ECMO设备在一些发达国家已广泛应用。在德国,平均每2万至4万人拥有一台ECMO。在美国,根据国际体外生命支持组织(ELSO)的数据,提供ECMO服务的医院数量从2008年的108家增加到2019年的264家。
两年前的疫情暴露全国重症监护设施不足短板。2020 年 5 月 20 日发改委公布了《公共卫生防控救治能力建设方案》,提出按照编制床位的 2-5%设置重症监护病床扩增 ICU 床位,ICU 配套医疗器械种类较多,包括监护仪、呼吸机、输液泵、除颤仪、心电图机等必选设备以及生化、血液分析仪、B超、ECMO 等可选设备。
经测算目前我国 ICU 床位数仅为 5.72 张/10 万人,仅就 ICU 必选设备,以文件中提到的 2%最低配置率,按照床位数 1:1 配置,测算出 ICU 扩容带来的增量空间至少有 172 亿元。若考虑到 5%的配置率以及可选设备,ICU 带来的增量空间更是高达数百亿级。

在 ICU 招标范围内,迈瑞份额保持最大,ECMO 被誉为 ICU 里把守“生死之门”的最后一道防线,未来如能成功研发出ECMO将进一步扩大自身的优势,造福广大的重症病患!
来源:器械之家


