






2022年上海医疗器械展会谈材料之聚碳酸酯基聚氨酯弹性体
2022-08-05
“聚氨酯”是聚氨基甲酸酯的简称,英文名称是polyurethane,英文缩写PU或PUR。它是一种有机高分子材料,被誉为“第五大塑料”。聚氨酯最早于1937年在德国发明,由异氰酸酯(单体)与羟基化合物(聚醚型或聚酯型)在一定比例下反应聚合而成。医用材料的头部企业们悉数参加2022年上海医疗器械展会Medtec China:韦恩堡、庄信万丰贵金属、田中贵金属(上海)、ELGILOY特种金属、三铃制线、江阴佩尔、麦迪斯、美国奥博锐、古河科技、沈阳中核舰航、路博润、NuSil、迈图、科思创、艾曼斯、塞拉尼斯、索尔维、龙海化工、江苏君华等。
聚碳酸酯基聚氨酯弹性体简介:
聚氨酯材料具有很好的生物相容性和血液相容性 ,加上它优异的力学性能、耐疲劳性和可加工性 ,一直以来都是备受重视的医用材料。聚碳酸酯基聚氨酯(PCU)材料最早开发于20世纪70年代,最开始主要应用在工业领域。而过去很多医用方面的研究主要集中在聚醚型聚氨酯方面,但大量的研究表面,聚碳酸酯基聚氨酯(PCU)材料在体内比聚醚型聚氨酯更耐氧化降解。因此,PCU材料在肿瘤科、心血管科、骨科等方面有更多的应用机会,被认为最具价值的医用合成高分子材料之一。
合成工艺:
1.本体聚合熔融
可按有无预反应分为预聚法和一步法。
· 预聚法∶将二异氰酸酯与大分子二醇先行反应一定时间,再加入扩链剂合成TPU。
· 一步法∶将大分子二醇、二异酸酯和扩链剂同时混合反应成TPU。
2.溶液预聚体法(两步法)
将二异氰酸酯先溶于溶剂中,再加入大分子二醇令其反应一段时间,最后加入扩链剂生成TPU。 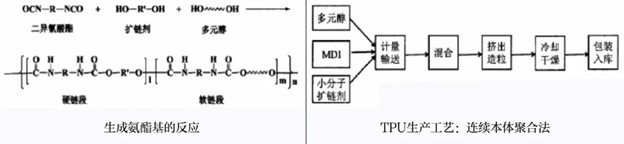
上游材料:
· 软段∶聚碳酸酯二元醇PCDL
· 硬段∶二异氰氨酸MDI
· 扩链剂∶丁二醇BDO
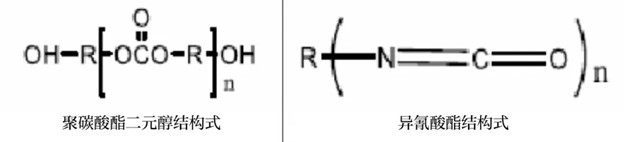
聚碳酸酯二元醇PCDL
特点∶结构规整、分子量分布窄,而且它不含酯键,耐候性能优异,与一般聚酯、聚醚系得二醇相比表现出相当优异的机械性能抗水解性、热稳定性、耐候性和抗化学品性能,同时还具有良好的生物相容性,是目前多元醇中综合性能较优异的品种。
二异氰氨酸MDI
特点∶价格低、挥发性较小,利于降低生产成本,而且符合工业安全防护和工人身体健康。缺点是易黄变。
内部结构特点:(硬段和软段)
热塑性聚氨酯弹性体的主链结构按照组成原料和性质不同,主要分为两部分,硬段部分和软段部分。其中,材料的低温性能主要来自于软段部分的特殊分子结构;材料的刚性和硬度主要来自于硬段部分的特殊分子结构。
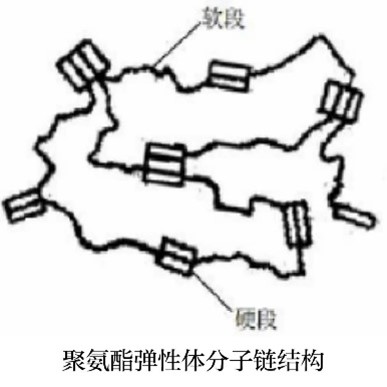
· 软段∶特殊分子结构主要来自于聚碳酸酯二元醇的酯基。由于酯基极性较强,因此聚碳酸酯基聚氨酯具有更优异的力学性能。
· 硬段∶特殊分子结构主要来自于异氰酸酯,它能给予材料一定的极性(氨基甲酸酯基团)和刚性(苯环),对提高其力学性能有利。
PCU性能特点:
一般聚氨酯弹性体的性能特点∶
1. 耐油性 2. 耐磨性优异 3.耐低温 4.耐老化 5. 弹性 6.耐化学性良好 7. 耐臭氧消毒
聚碳酸酯基聚氨酯:
· 与传统型多元醇所合成的聚氨酯材料相比,聚碳酸酯型聚氨酯具有更好的力学性能、耐水解性、耐热性、耐氧化性及在高温或潮湿等情况下具有长期稳定的绝缘性质。
· 软段由聚碳酸酯链段构成,在生理环境下,碳酸酯键比醚键更加稳定。因此,聚碳酸酯基聚氨酯更耐血液中巨噬细胞所产生的氧自由基降解。
· 用TMP作为扩链剂合成的PCU,具有优良的形状记忆性能。同时,增加硬段含量,减少软段含量,有利于材料的形状固定,而软段也能表现出较好的形状回复率。在生物医学领域可以有更多应用机会。
· 聚碳酸酯聚氨酯的水蒸气透过率比聚醚型聚氨酯低2至4倍。
此外,2022年上海医疗器械展会Medtec China了解到聚碳酸酯基聚氨酯通过调整如下配方,可以提高抗凝血性∶
· 表面改性(引入疏水性或亲水性侧链如聚乙二醇/聚酰胺、引入生物活性大分子)
· 合成微相不均匀结构
· 调整软硬段比例
· 改变扩链剂的类型及含量
微相分离对性能的影响
微相分离对性能的影响∶
聚氨酯与其他生物材料相比,主要的物理结构特征是微相分离结构。聚氨酯的物理性质不仅与其化学结构有关,而且与微相分离的程度有关。一般地,两相分离越完善,对材料的力学性能和耐热性能越有利。
微相分离也可称之为两相分离,两相指的是由硬段形成的硬段相和由软段形成的软段相。软段和硬段的分子结构不同、所含基团的极性和刚性不同,在热力学上具有自发分离的倾向,即不相容性。硬链段很容易聚集在一起,形成许多微区,分布于软段相中,这种现象叫微相分离。
硬段∶含有强极性基团(氨基甲酸酯基)和刚性基团(苯环),容易聚集,形成规整性好的结构,类似棒状的伸直状态。
软段∶只有几乎没有极性的基团(醚键)和极性相对较弱的基团(酯基),结构规整性很差,聚集成杂乱无规的线团状态。
2022年上海医疗器械展会Medtec China技术论坛A:医疗器械诞生的助推器——创新医用材料/配件及精加工(一)议题覆盖医用塑料在医疗器械生产过程中的应用、汉高医疗行业创新解决方案、埃万特医疗级聚合物解决方案如何为您的体外诊断(IVD)应用增加价值、蔻兰多彩高性能材料解决方案在医疗行业的应用等。点击快速预登记。
软段和硬段比例的对性能的影响
硬段含量是指硬段质量在聚氨酯弹性体中所占的百分比。它是配方计算和分子设计的一个重要参数,通过它可以调节聚氨酯弹性体的性能(如强度、硬度、拉断长率)。硬段含量越高微相分离概率越大。
· 硬段含量小于10%∶由于硬段含量太低、硬段间作用力太弱硬段微区无法形成,此时硬段溶于软段中形成单相,使得弹性体材料只表现出单一软段相的性能;
· 硬段含量在25%~40%∶硬段含量变高,硬段间作用力增强,虽然此时硬段相已具雏形,但软段相仍为连续相,此时的弹性体材料软段相性能较突出,如弹性好、强度差;
·硬段含量在40%~60%∶硬段含量继续提高,此时的硬段相区既可以作为连续相,又可以作为分散相,两相分离程度相对较高,材料的性能较优异。
· 硬段含量大超过60%∶两相发生了逆转,此时的硬段相为连续相,材料主要表现出硬段的性能,如力学强度高,低温柔顺性和加工性能相对较差。
来源:东莞市富临塑胶原料有限公司


