






高端医疗设备展解密微流控制备最新研发技术突破与成果!
2022-06-07
微流控(Microfluidics)是对体积在纳升到微升的微量流体进行精确操控和分析的技术,又称为芯片实验室(Lab on a chip)。这种技术可实现采样、稀释、加试剂、分离、检测等实验流程以及各种生化反应在几平方厘米或更小的芯片上的集成,从而减少样品试剂消耗、提高检测灵敏度、缩短反应时间、降低平均成本。经常应用于IVD行业中,包括生化分析,免疫诊断和分子诊断。微流控管路结构在微米至纳米尺度,流体在此尺度下展现与宏观尺度不同的特征(例如:层流与微液滴)。利用微尺度下流体特征,微流控可以完成一些常规方法难以取得的微操作。与传统分析技术相比,微流控技术具有以下优势:样品与试剂消耗量少、能耗低、灵敏度高、可大量平行处理样品、可实现设备的集成化、微型化和便携化。参加高端医疗设备展Medtec中国展的许多工程师也表示微流控已经发展成为医学、材料等学科的交叉前沿研究热点,学术界和工业界都备受关注。
关键问题
目前,可用于制备微流控的技术和材料有限,阻碍了微流控的广泛应用与发展。现有的各种技术各有优缺点。光刻蚀技术可在硅、玻璃、陶瓷等硬质材料中制备高精度微流控芯片。在通过光刻蚀等微加工技术获得主模板后,软刻蚀技术采用可固化高分子(例如PDMS)进行模板复制,该类高分子成本较高,且工艺流程复杂。通常,这两种技术制备通道的顶部为敞开状态,需要额外步骤进行封闭。而封闭环节增加工艺流程,也可能会引发加工故障。纸质微流控(paper microfluidics)是近期另一研究热点,有低成本优势,但纸质微流控的缺点是难以对内部孔径进行精确调控,通道制备精度较低。微流控亟需从制备技术层面进行突破,以简化工艺流程、降低材料成本(适用廉价材料)、提高精度、丰富功能为目的。
新思路

2019年,京都大学的Sivaniah研究组开发出规整微纤维(Organized Microfibrillation, OM)方法。有别于常规光刻蚀方法,OM方法在光敏高分子材料中引入光学驻波进行分层交替交联,然后采用弱溶剂在材料中构建出尺寸规则的微纤维和孔。微孔层周期性排列,对特定波长入射光产生布拉格反射,形成结构色彩效应。在此基础上,Sivaniah组研究员秦德韬(Detao Qin)博士运用OM方法开发出自带结构色彩的微流控制备技术,可在厚度仅为1微米的高分子薄膜中高精度打印出自封闭微流控通道,相关论文近日发表在Nature Communications上。
技术方案:
OM微流控的制备方法:驻波光刻步骤采用掩模工具在曝光区域印制流体通道的图案,然后通过弱溶剂显影形成多层微孔的自封闭通道。 技术优势:
高端医疗设备展Medtec中国展多年来致力于整合全球医疗技术及资源,关注医疗健康关键核心技术突破,认为可以在普通工程高分子(例如:聚苯乙烯、聚甲基丙烯酸甲酯)中高精度制备出亚微米孔的自封闭通道。自带结构色彩功能,可用于原位检测。工艺流程较为简便 (仅需要铸膜、光刻和显影3个步骤)。
技术细节
OM微流控的制备
OM微流控的制备需要掩模工具。普通打印机打印OHP薄膜可用作低成本掩模,方法简便,但精度仅为毫米级。采用OHP薄膜和普通LED光源在PET、玻璃、硅片等基底上制备出OM微流控装置见图1c-e。
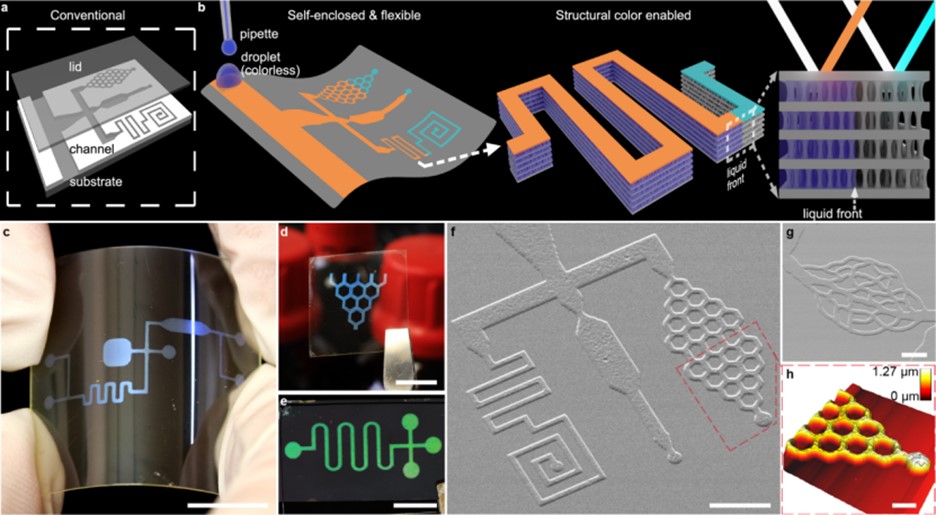
图1. 规整微纤维方法打印出结构色彩型薄膜微流控。比例尺:图1c-e, 1 cm;图1f, 50μm;图1g, 10μm;图1h, 20μm。
MicroLED光刻设备可用于OM通道的高精度制备,通道微打印精度可到5 μm (图1f-h)。前期工作(Nature, 2019, 570)中,OM微打印局限于不透明基底(硅片)。本论文突破这一局限,在透明基底实现微打印。透明基底拓宽了OM的应用,特别是,生物医学领域观测微流控常用倒置显微镜,需要微流控芯片有透光功能。液体可通过毛细作用力进入OM通道。流体实验表明,OM通道的内部微孔在平面内连通,液体按印制图案进行流动(图2)。
高端医疗设备展Medtec中国展涵盖更多高精尖技术,“技术论坛C:精密加工设备与技术应用论坛” 将于8月31日下午在上海世博展览馆现场开讲!专家将对飞秒激光助力极端制造与超精密加工等精密加工技术进行案例分享,点击预登记即可免费参与会议!

图2. 共聚焦显微镜观测液体在OM通道的流动。基底:硅片(图2a,b)和玻璃(图2c,d)。图2a-c: 荧光分子为ATTO 495 (绿色)和ATTO 610 (红色)。图2d: 绿色荧光蛋白。比例尺:图2a,b, 100 μm;图2c, 500 μm;图2d, 20 μm。
OM微流控的结构色彩特性
OM通道内部是亚微米孔的多层堆叠,微孔孔径(20-200 nm)决定流体动力学特征。同时,孔径与多层结构的层间距正相关,因此,OM微流控的将结构色彩与流体力学特征相耦合。图3a,b表明OM流体力学特征不取决于打印通道的表观宽度。图3c,d进一步表明,液体在OM通道的扩散参数(dL2/dt)和结构色布拉格峰位正相关。OM技术具备将流体力学性能“可视化”特征:结构颜色越红,表明通道内部孔径越大,液体的扩散速率越快。
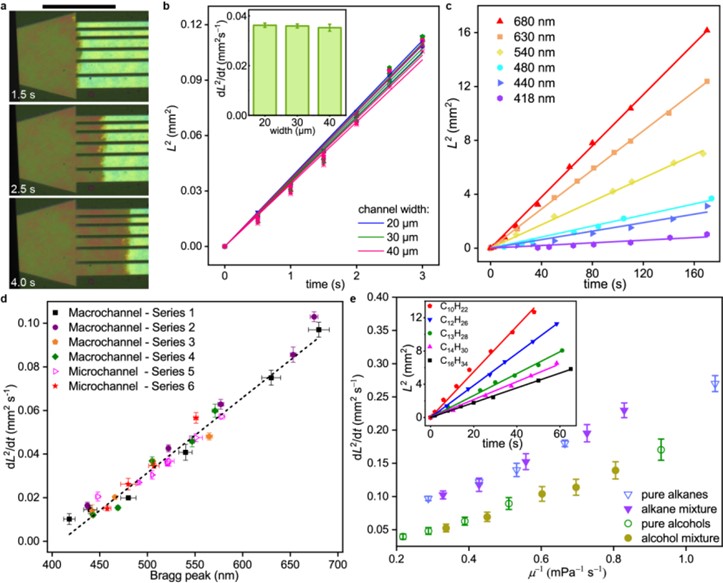
图3. OM微流控的流体动力学特性。比例尺:图3a, 200 μm。
OM通道的折射系数会因为液体流入而发生改变,这导致结构色彩发生变化(图4a)。OM通道布拉格反射峰的峰位和峰高会对流体的折射系数产生响应,因此,OM微流控可基于结构色彩进行原位检测(图4b-d)。图4e展示OM微流控薄膜透明特性。
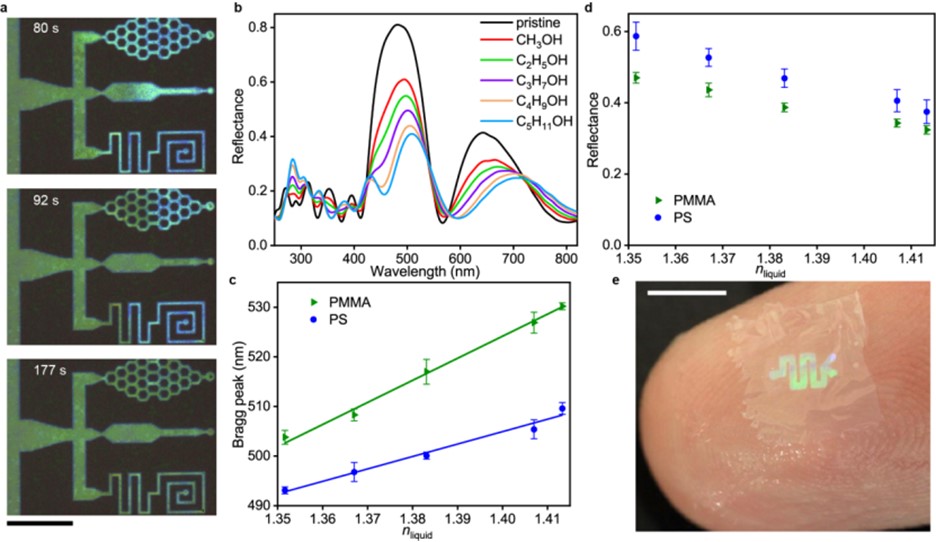
图4. 基于结构色彩的原位检测。比例尺:图4a, 100 μm;图4e, 0.5 mm。
OM微流控的筛分功能
OM微流控可针对特定目的对微孔尺寸进行精调。通过控制区域曝光能量,可在OM单芯片中串联微孔差异的多级通道。如图5a-e所示:OM主通道区域采用高曝光能量,而侧枝通道区域采用低曝光能量。该方法制备OM芯片主通道的结构色比侧枝通道更红。通道截面的扫描电镜照片显示主通道的微孔孔径更大(图5f-h)。

图5. OM微流控内部孔径调控。图5c-e: (1) 表示OM主通道,(2) 表示侧枝通道。比例尺:图5b, 200 μm;图5c-e, 100 μm;图5f, 50 μm;图5g,h, 200 nm。
上述微孔分级串联通道被用在水系流体中筛分生物分子。分子量不同的葡聚糖(蓝色荧光:3-kDa 葡聚糖,红色荧光:70-kDa葡聚糖)筛分结果见图6a。葡聚糖-蛋白质(蓝色荧光 :3-kDa 葡聚糖,红色荧光:27-kDa 红色荧光蛋白)筛分结果见图6b,分离效果更好。近期研究热点显示SARS-CoV-2病毒和糖尿病的病理学存在关联,图6c展示OM通道对胰岛素(Insulin, 6 kDa, 红色荧光)和SARS-CoV-2病毒核衣壳蛋白(Nucleocapsid protein, 55 kDa, 绿色荧光)的筛分结果。实验显示,OM微流控对不同分子量蛋白质的筛分效果要优于葡聚糖的筛分。其原因可能为蛋白质分子量为明确的定值,而葡聚糖自身则存在分子量分布。
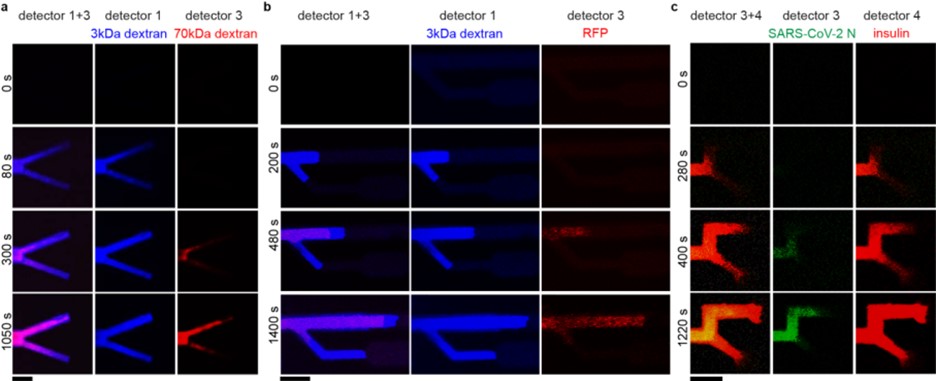
图6. OM微流控筛分生物大分子。共聚焦显微镜观测结果,生物分子通过荧光区分。比例尺:100 μm。
展望
OM微流控技术可高精度打印出多层亚微米孔的自封闭通道。OM薄膜厚度仅为1微米,是目前各技术中最薄的微流控芯片,有助于跨膜物质传输方面的应用。该技术工艺流程简单,适用普通工程高分子(例如:聚苯乙烯,聚甲基丙烯酸甲酯),可制备柔性、透明等多样式微流控芯片。
微流控系统实现了小型化,不需要额外的设备,离主流商业应用更进一步,其框架概念可广泛应用于医疗诊断。该技术有望为医药测试、皮肤接触装置、生化检测等技术的升级发展打开新局面。
来源:纳米人


