






医疗器械耗材粘接方案
2021-08-19
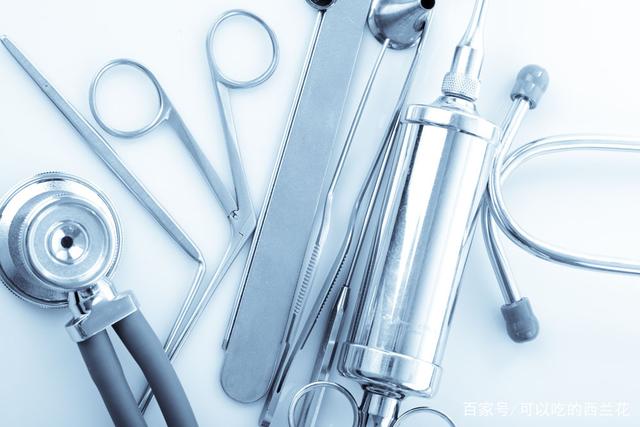
哪些医疗器械及耗材粘接密封使用UV胶?
医疗器械用UV胶水进行粘接,使得自动化装配更加容易,医疗UV胶水能在几秒钟内完成固化。UV胶行业内CRCBOND医疗级UV胶是一种单组分医疗级紫外光固化胶粘剂,通过VSPVI级认证和ISO-10993生物相容性测试;收缩率低,塑料粘接不会产生应力开裂;柔韧性配方,耐冲击、耐振动效果极优;粘度适中,可用于手工或点胶机施胶。
生物相容性测试;收缩率低,塑料粘接不会产生应力开裂;柔韧性配方,耐冲击、耐振动效果极优;粘度适中,可用于手工或点胶机施胶。
哪些医疗器械及耗材粘接密封使用UV胶?
目前主要应用在这些医疗器械及耗材上:1.麻醉面罩、2.注射器、3.导液管4.静脉输液管、5.血管植入配件、6.内窥镜、8.管状排水装置、9.气管管道、10.血液氧合器、11.助听器、12.生物芯片、13.探测,监控,以及图像器械、14.粘接PVC, 热塑料(聚碳酸脂据和ABS)等等……
四种医用塑料粘合剂解决方案
以下是5-6月发行的MPMN中强调的四种医用粘合剂技术:
1. 光感型粘合剂
Dymax的光固化Encompass技术结合了该公司的专利——See-Cure 变色技术和红外荧光技术,形成了一种单色光固化产品。Dymax(托灵顿,康涅狄格)给一次性医疗器械制造商提供用于单支导管粘合的粘合剂,这种粘合剂通过发光二极管激发,再用该公司的红外荧光技术和专利技术See-Cure把它固化。这种粘合剂在未固化状态时呈蓝色,完全激发时变为无色;然后,在365纳米波长下的低强度黑光检查时,它发出鲜红的荧光,以便轻易的确认粘合剂精确的放置在导管上。制造商的无溶剂接缝和通过美国药典六级以及ISO 10993标准的生物相容的光固化型粘合剂被设计用于快速固化和加工医疗设备装配。这些技术能接受γ、环氧乙烷和电子束灭菌技术。

光感型粘合剂
2. 组合粘合剂
由Henkel公司(落基山,康涅狄格)开发的两种柔韧的速粘剂,Loctite 4902 和Loctite 4903,它们分别提供了155 %和86 %的伸长率。他们可以在几秒钟内提供高强度的粘合。如同丙烯酸和尿烷粘合剂那样灵活,这两种氰基丙烯酸酯粘合剂能粘合塑料、橡胶、金属和其他材料,并提供符合ISO 10993标准的生物相容性。它们适合用在一次性医疗器械的组装中。单质低粘度的粘合剂容易流动到任何基质上,通常5秒内就能固定好组件。更柔韧的一种粘合剂拥有57,900 psi的模量, 伸长率达到85 %,表现出78 100psi的模量。这两种粘合剂都是设计用于贴身固定柔韧部件,并且可以容易地粘合不同的基底,包括塑料和弹性体。对难以粘合的基底,引物能增强粘合强度。
3. 紫外灯固化粘合系统
Optics公司(马萨诸塞州沃本)发布了新发明,用于工业UV固化的Aurora UV Classic Line Sources,该发明用来进行粘合剂、涂料和密封剂的光聚合。Aurora Classic的光学设计专利提供了远距离的峰值辐射,该辐射来自发射窗口,在二维平面中均匀分布。该系统适用于UV固化,这种情况下机器框架或复杂零件轮廓能防止靠近固化表面的UV二极管被粘起来 。该系统适用于从4-24英寸的五种光束长度,以及四种标准UV二极管中心波长,包括365 、 385 、 395 和405纳米。Aurora UV Classic Line Sources可以在自然对流冷却下工作。它还提供了风扇冷却增压这种选配来驱动线光源,将峰值辐射提升了两倍。

紫外灯固化
4. 光固化粘合剂
Ellsworth Adhesives(威斯康辛日耳曼)是一家粘合剂、特种化学品和点胶设备的全球分销商,该公司为医疗装置应用,如针接合,搜集装置和换能器壳体组件提供Henkel Loctite 3211。Loctite 3211是光固化型丙烯酸粘合剂,它能以最小的开裂应力迅速形成柔性接头。它适用于玻璃、金属、聚碳酸酯、热塑性塑料。Loctite 3211的灵活性加强了粘合区域的承载和减震特性,而不会造成常规模型应力水品的应力开裂。当暴露于足够强度的紫外光或可见光时,Loctite 3211迅速固化形成柔韧透明的粘合。Loctite 3211的触变性降低了液体制品应用到基底上的移动。Loctite 3211通过了ISO 10993认证。
来源:可以吃的西兰花


