






血管介入手术机器人,冠脉介入、神经介入等国外公司进展 | 血管介入系列02
2022-02-28
冠脉介入机器人
冠脉是介入手术机器人的主战场,目前绝大多数的产业化都集中于这个领域。
与上篇讲的Magellan应用不同的是:
1. 冠脉机器人主要操控极细的导丝(直径约0.9-0.3毫米不等)在极细的冠脉中使用,而Magellan即使也能操控同样尺寸的导丝,但是它的主角是自家的可弯曲导管(直径都多在2毫米以上)在较粗血管中使用。
2. 冠脉介入机器人一般都整合了冠脉球囊和支架的放置,于是能够进行一定的治疗任务。这里要指出,机器人仍然只能“递送”这些微导管,而球囊、支架放置由医生手工完成。而这类机器人大多都能整合现有市面上的一些介入器械,使机器人更像是使用这些器械的“平台”(这点跟骨科机器人类似)。从而我们可以看出Magellan机器人即使使用了昂贵的黑科技导管,但是也只能完成介入手术的前一部分任务,也许是它难以推广的一个原因。
最有名的冠脉介入机器人莫过于Corindus的Corpath机器人了(现已被西门子医疗收购)。Corindus公司在2002年就成立了,或许比Hansen要早,可以说是商业化血管介入机器人的鼻祖。(此前思宇医械观察介绍过Corindus,链接如下:
西门子新欢,飞利浦旧爱,血管机器人公司Corindus如何在巨头间游走)
2012年上市的第一代Corpath 200机器人看上去非常简单:
一组摩擦轮来递送导丝,一个机构旋转导丝,另一组摩擦轮来递送球囊、支架导管。
它的定位十分明确——在冠脉中操作导丝和治疗用的微导管。它似乎发挥了之前提到的减少辐射的优势,同时能让医生在更为惬意的条件下完成这个繁琐困难的介入任务。
第二代产品于2016年面世至今的Corpath GRX系统,它主要的改进是增加了小范围操作粗导管的功能,另外在被西门子收购前(或许不是巧合)非常活跃地开发了长距离远程手术、自动手术模块等功能。目前资料最丰富,应用最广泛的介入机器人平台非Corpath莫属,而随着医疗机器人的热潮,也在2021年在海南进行第一例临床,开启了进入中国的大门。
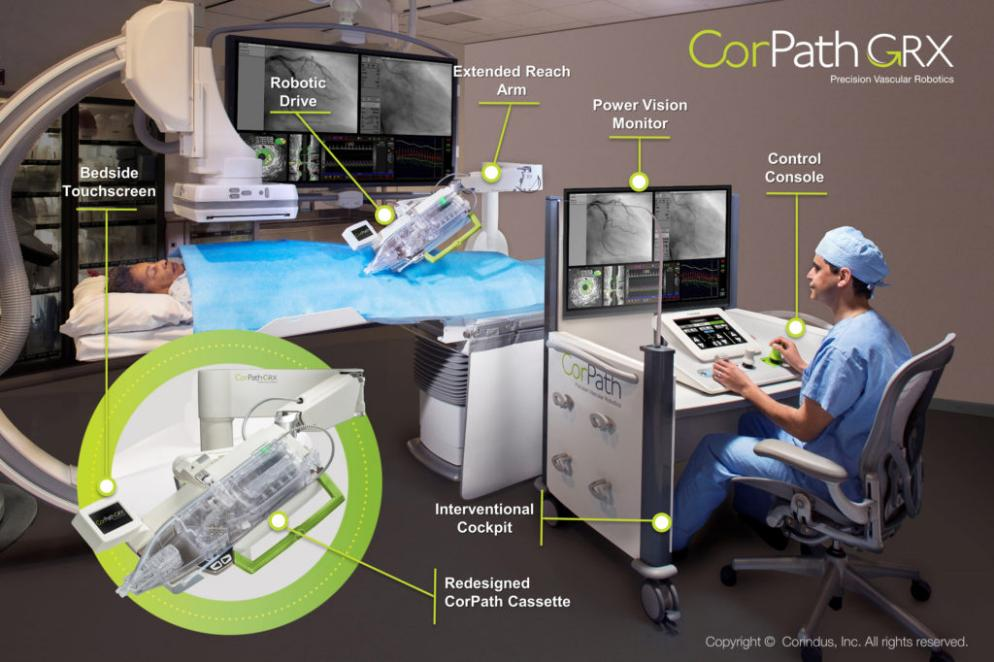
▲西门子Corindus CorPath GRX 机器人
另一台已经上市的机器人是法国RobotCath的R-One机器人。它面世之初看上去像是Corpath 200的翻版。最大的区别是导丝的递送机构采用了夹爪递送的模式(跟摩擦轮相反,一般递送的机构莫过于这两种)。
目前上海微创机器人和RoboCath在华合作成立知脉公司,引入R-One血管介入机器人。2021年11月下旬在301医院完成首例中国注册临床试验;并已经做好了培训系统的准备。
RobotCath R-ONE®在中国完成的首例注册临床试验
西门子医疗介入手术机器人捆绑虚拟患者系统2022年在中国销售

▲RoboCath R-one机器人
神经介入手术机器人
神经介入机器人更像是一个未来发展方向,同样是在极微小的血管内操作导丝和微导管,随着神经介入器械自身的快速发展,神经介入变成手术机器人的兵家必争之地。
在Magellan退市的前一年,最后的一波技术更新可圈可点,包括整合第三方微导管、4Fr超微可扭转导管和神经介入的应用申请,似乎Hansen最后的救命稻草就是神经介入。
Corindus Corpath GRX面世不久就扎进了神经介入,并于2019年得到了CE认证,但是就早期的临床报告上说,Corpath硬件和软件上并没有对神经介入进行太多的优化,而且缺乏对微导管(直接1mm左右)的支持。相信Corpath正在进行对神经应用专项的升级。
还很难被定义的介入手术机器人
以色列的Microbot Medical公司在2020年初公布了他们的Liberty介入机器人系统。(进展见MedRobot文章
血管介入机器人 | Microbot Medical 开始在美国为 LIBERTY® 机器人系统寻找临床站点 | 临床)
这个机器人非常具有创新性。据称它是世界上第一个一次性手术机器人。它的构造有点像一个老式的磁带,特制的导管提前卷好塞入机器人内,自研的导管头部可以主动旋转(使用了一个很简单的方式)。于是它的体积很小,结构似乎也不复杂,所以成本可以很低。
然而目前披露的信息依旧不多,而且它所针对的介入临床应用似乎也不够清晰。但是随着一次性医疗器械的火热发展,这种低成本、轻量化的介入手术机器人很可能也在未来有一席之地。
来源:MedRobot


