






Medtec 中国展关注CT成像:光子计数 CT或成下一个时代?
2022-05-07
光子计数 CT (PCCT) 是目前最有前途的一种UHRCT技术,该技术可获取更高空间分辨率的图像, 多年持续关注着研发&制造等前沿技术,在与传统 CT 在性能方面的对比后,Medtec 中国展发现这种新模式不仅具有更好的软组织对比度和空间分辨率,同时也减少了噪声、光晕和射线硬化伪影。或许冠脉CTA已经成为侵入性冠脉造影的一个替代诊断方案,尤其是在冠心病的阴性预测方面。
但钙化或支架伪影会影响病变的准确评估。近来CT 硬件和最新重建技术的发展使 CT 血管造影具有超高空间分辨率 (UHRCT),可克服钙化、支架或小血管病变在当前传统CT中可视化和量化的局限性(图1)。
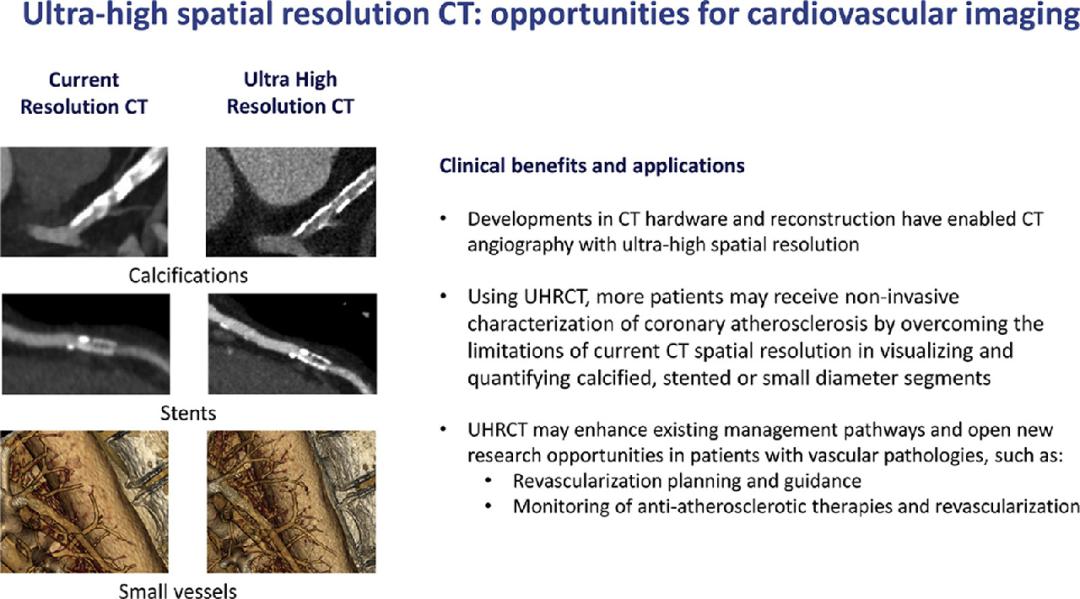
图1. UHRCT与传统CT相比,钙化、支架内病变和小血管的造影图像
近日,Medtec 中国展注意到,第一个PCCT系统已经商用,采用基于碲化镉的光子计数探测器的双源系统,具有标准多能模式和UHR模式两种采集模式。在 UHR 模式下,理论上可以实现 0.125 mm 的平面内分辨率,具有 120 × 0.2 mm (24 mm) 的 z 覆盖和 1024 × 1024 矩阵重建(图2)。
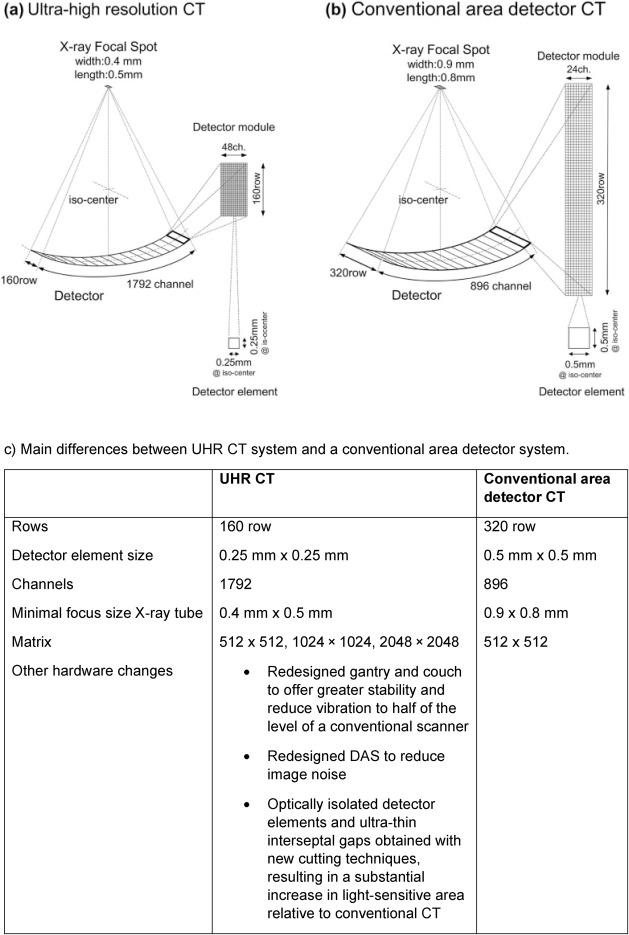
图2. 专用 UHRCT 和传统高端 CT 扫描的比较。(A):在 UHRCT 系统(Aquilion Precision)中,等中心处的探测器元件尺寸为 0.25 × 0.25 mm,探测器有 160 排1792 个通道。X射线管的最小焦点尺寸为0.4×0.5mm。(B):在传统的高端广域探测器CT(Aquilion ONE)中,等中心处的探测器元件尺寸为 0.5×0.5mm,探测器有320排 896个通道。X射线管的最小焦点尺寸为 0.9×0.8mm。(C):与传统区域探测器 CT 相比,表格提供了专门针对 UHR CT 包括硬件设计的主要区别 Motoyama 等人报告了第一个UHRCT临床观察结果。与传统 CT 相比,UHRCT 上的钙化更小,伪影更少,显著减少了传统CT上被诊断为 ≥50% 狭窄的钙化病变。与标准分辨率 CT 相比,UHRCT 上的支架管腔更大,支架梁更明显,也更薄(图3)。大多数直径 ≥ 2.5 mm 的支架可以通过 UHRCT 来进行评估,不过直径≤2.25 mm 支架仍然存在挑战。
图3. 既往支架植入患者的UHRCT成像与重建。前降支UHRCT多平面重建(A、E)和横截面图像(B、C、D)上,支架远端新内膜增生以及其远端边缘周围的斑块可以清晰分辨。这种细节在模拟的正常分辨率图像 (F) 上就不那么明显了。在3D 渲染图像(G、H、I)上,可以看到冠脉包括远端支架边缘的管腔变窄(I,箭头)的逼真影像。
UHRCT 系统与侵入性的冠脉造影(ICA)之间诊断准确性的比较,尽管数据和样本量还不大,但已经初露曙光。一个对59例患者、平均钙化积分达171分的对比研究结果显示,与ICA相比,UHRCT 检测到严重狭窄的敏感性和特异性达 100% 和 80%;Takagi 等人研究了 38例患者的对照,结果UHRCT敏感100%,特异性67%。
最近一项研究对15 例重度钙化(平均钙化积分 1205 )且大多数达到肥胖标准的患者进行了UHRCT 检查显示,所有图像均可判读(图4)。平均图像质量和诊断置信度得分(按 1-5 分排名)分别为 4.1 ± 0.8 和 4.3 ± 0.9。与 ICA 的比较敏感性为86% ,特异性达88% 。重要的是,尽管钙化积分非常高,UHRCT 仍能排除阻塞性冠脉疾病(也就是直径狭窄≥50%)。
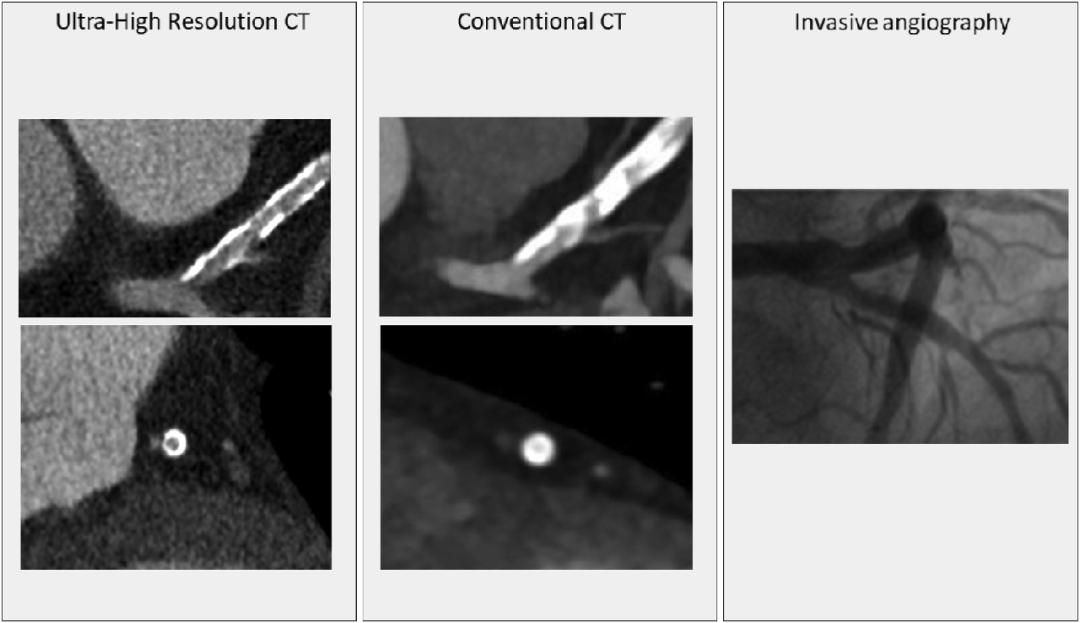
图4. 这是一例钙化积分超过 2000 的患者 UHRCT 与常规 CT 的冠脉影像比较。在 UHRCT 中,尽管前降支存在广泛严重钙化,仍可以排除前降支近段存在重度狭窄。而常规 CT 上钙化导致的严重伪影无法确定前降支是否有重度狭窄。侵入性的冠状动脉造影证实前降支没有明显的狭窄
【展望】
UHRCT有可能今后代替冠脉造影,成为冠心病首选的影像学诊断手段。未来UHRCT的应用将会进一步减少侵入性冠脉造影的比例。结合CTFFR、三维重建,来指导冠心病的治疗,包括药物和血运重建术前策略的制定。
Medtec 中国展多年深耕医疗器械市场,2022年Medtec 中国展高端有源医疗设备核心部件与技术论坛涵盖高端影像设备的应用以及CT等技术的发展,如果您目前主要从事心血管病的影像诊断及介入治疗工作,点击立即获取参会机会。
来源: CCI心血管医生创新俱乐部


