






白内障克星-人工晶体进化史和赛道分析
2020-11-26
每两个盲人中,就有一位是白内障致盲的。
据2015年WHO的调查估计,白内障占致盲眼病的51%,全世界由白内障致盲的患者约有2000万。
那什么是白内障?简单说,白内障就是指晶状体混浊,阻止光线进入眼睛。晶状体是人眼中一个“透明状体”,位于角膜和瞳孔后方。光线需要通过晶状体的折射聚焦到视网膜上,当晶状体混浊时会影响到光线折射过程,使事物看起来模糊朦胧。
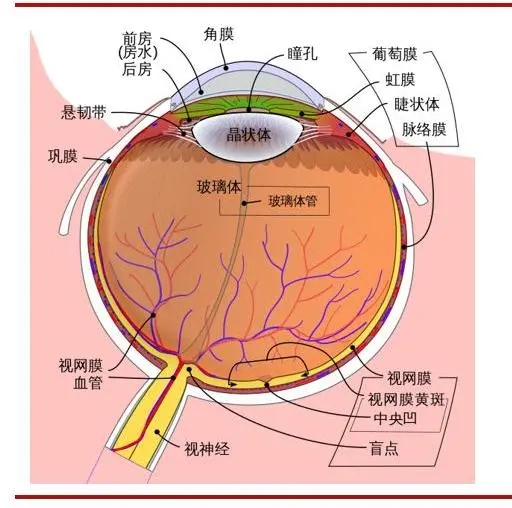 (图:眼睛结构解剖图)
(图:眼睛结构解剖图)
(图片来源:维基百科 思宇医械观察整理) 白内障有多种类型,最常见的是老年白内障,属于年龄相关性疾病,岁数大了就容易得这种病。糖尿病、长期日光照射、吸烟和肥胖均是老年性白内障的相关风险因素。此外,也有儿童天生患有此病,以及部分患者在炎症、受伤和其他疾病后出现白内障。
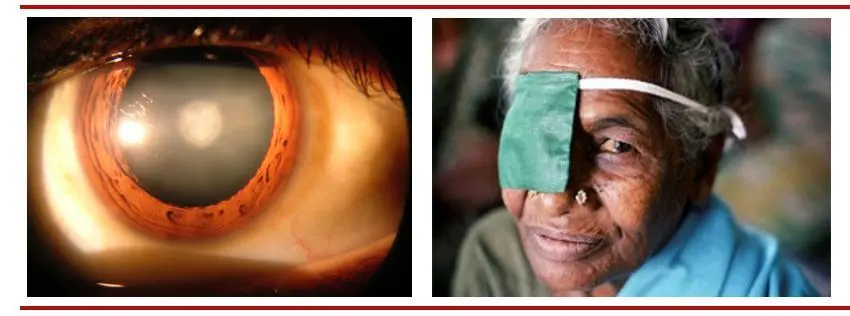 (图:白内障患者的眼睛晶状体混浊(左)白内障患者(右))
(图:白内障患者的眼睛晶状体混浊(左)白内障患者(右))
(图片来源:维基百科与WHO官网 思宇医械观察整理) 白内障手术及人工晶状体植入术 手术是白内障唯一有效的治疗手段。通过手术可摘除已经混浊的晶状体,然后植入人工晶体使视力恢复。白内障手术术式已经历两代进化,第一代是囊外白内障摘除手术,其并发症多且不易植入人工晶状体,此旧式的手术已很少使用。
第二代是主流的白内障超声乳化术,手术过程较简单,首先在角膜侧面切一个小切口,使用超声设备或激光将混浊晶状体分成小碎片,然后从眼睛上轻轻吸出小碎片,最后将人造晶状体牢固地插入瞳孔后方,使其与自然晶状体占据的位置相同,切口无需缝合即可自行愈合。白内障超声乳化术目前已比较成熟,手术过程通常在15分钟内就可完成。
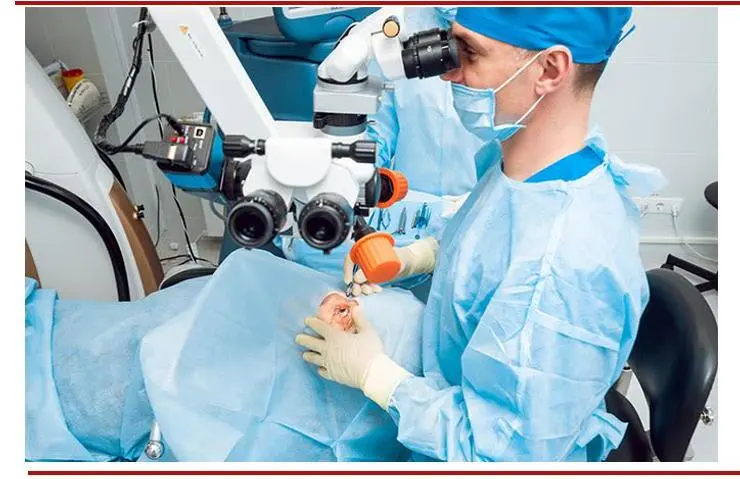 (图:白内障手术)
(图:白内障手术)
(图片来源:allaboutvision网站 思宇医械观察整理)
人工晶状体的重要地位及进化史 人工晶状体(IOL)是治疗白内障必不可少的医疗光学器件,在摘除混浊晶状体的白内障手术后,如果不植入IOL,将会产生更加严重的屈光不正,这将严重影响病人的生活。
作为植入性器件,IOL是永久固定在眼睛内部的,不会掉落也无需清洁,患者几乎不会有任何感觉。更重要的是,IOL不像其他植入性物质那样被组织排斥,植入晶状体的部位无排斥性组织,因此可使其可以长久存在。
1949年,哈罗德·里德利爵士于伦敦圣托马斯医院,为白内障病人手术后,实现了第一例人工晶状体植入术,当时IOL的材料还是聚甲基丙烯酸甲酯(又称亚克力),它是一种坚硬的不可折叠材料,因此眼科医生在植入这种硬性人工晶状体时极具挑战性。
 (图:亚克力(高透明材料,多用作玻璃替代品))
(图:亚克力(高透明材料,多用作玻璃替代品))
(图片来源:百度百科 思宇医械观察整理) 1980年左右,爱泼斯坦使用硅树脂制成了可折叠软性人工晶体。这使得眼科医生可以通过3毫米的小切口植入IOL,相比于不可折叠的IOL需要 5-7毫米切口,可以说是得到了很大的提高。1990年,第一个被FDA批准的可折叠人工晶体是Allergan公司的硅胶镜片人工晶体,可折叠IOL已成为现在最普遍植入的镜片。
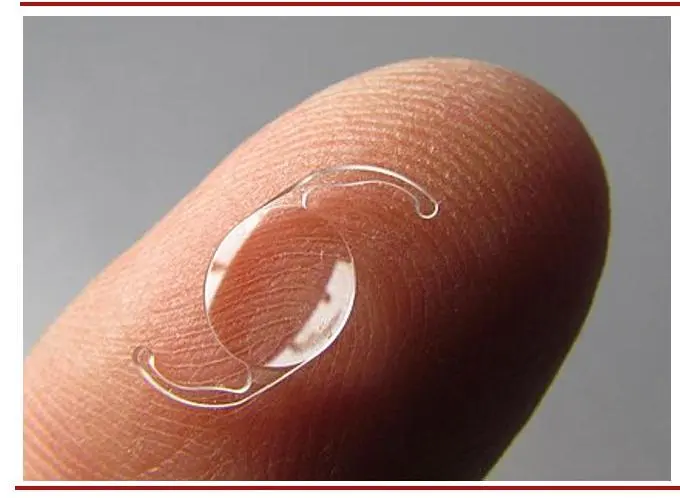 (图:后房型人工晶状体)
(图:后房型人工晶状体)
(图片来源:维基百科 思宇医械观察整理)
人工晶状体分类介绍及选择
现代人工晶状体的设计是美国眼科医生史蒂芬·希灵博士创造的,他创新性设计了三件式模型,把圆形光学器件与两个柔性支柱触觉件融合在一起,柔性支柱触觉件的作用就像张力弹簧一样,可以自动将植入眼球的晶状体置于眼室的中心位置。
从人工晶状体植入位置上讲,可分为前房IOL和后房IOL,后房通常是人工晶体的首选位置,因为在白内障摘除手术后,留下了透明的具有支撑性的晶状体囊袋,它提供了一个放置永久性人工晶体植入物的最佳位置。并在植入IOL后收缩从而将其固定,其位置与原天然晶状体相同。前房性IOL目前不太常见,仅在患者无法选择后房性IOL的情况有时才会使用。
从镜片焦点方面讲,人工IOL可分为单焦点镜片、多焦点镜片和散光镜片。目前单焦点镜片仍是最多见的植入类型。“单焦点”是指它可提供一个聚焦清晰的视觉,大多数选择单焦点的患者会选择远距离视力,然后通过佩戴眼镜来辅助近距离视觉任务,比如阅读和看电脑手机。
多焦点人工晶状体的“多”意味着不止一个焦点,它同时提供了提供远距视觉和近距视觉,使患者在看远处和近处事物时均可清晰,减少患者对眼镜的依赖。
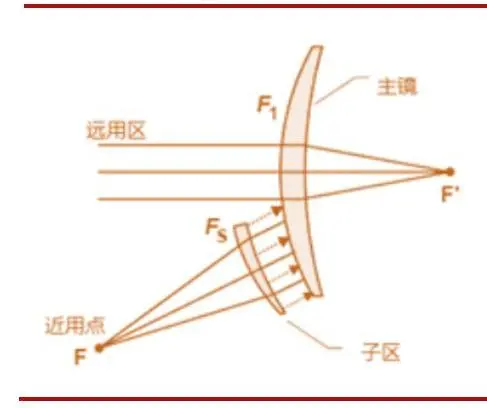 (图:多焦点示意图)
(图:多焦点示意图)
(图片来源:网络 思宇医械观察整理)
此外还有散光透镜可以选择,可用来矫正散光的单焦点透镜,减少患者术后配戴眼镜矫正散光的需要。
赛道分析 尽管可折叠软性晶状体成为手术植入主流,但在中国,仍有30%的白内障患者使用硬性晶状体(低端人工晶状体),这主要是价格低原因。因为其不可折叠,会使得手术切口增大一倍以上,达到6毫米。剩下的70%患者使用的软性晶状体大多是从国外进口。
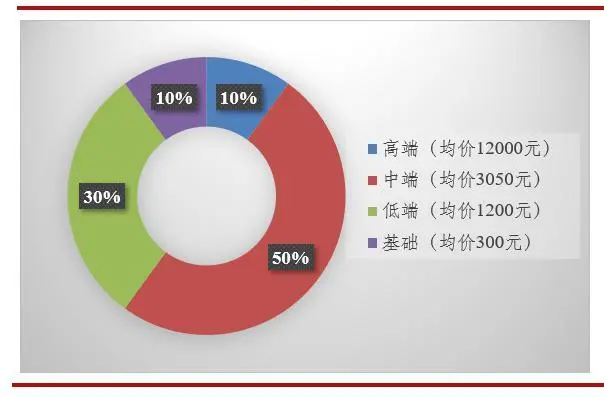 (图:2019年我国人工晶体各梯队市场份额)
(图:2019年我国人工晶体各梯队市场份额)
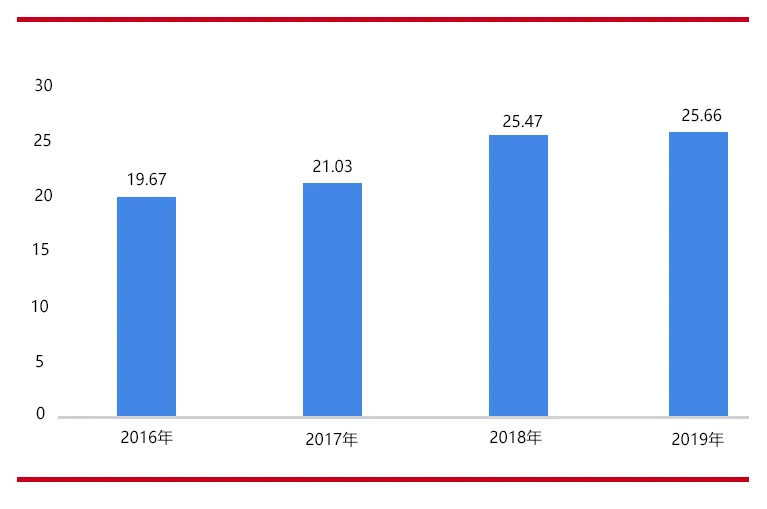
(图片来源:爱博医疗数据 思宇医械观察整理) 据估计全国有年龄在60岁以上的1.3亿潜在白内障患者,目前需要白内障手术的患者数接近500万,且每年以40万人的速度增加。2019年,我国人工晶状体市场规模约25.66亿元。IOL作为三类医疗器材,属于高值医用耗材,发展中国自主品牌的IOL必要性很强,这可打破国外品牌的垄断和溢价能力,降低国家和社会的经济负担。
目前IOL巨头由四家国际公司组成,美国的爱尔康、强生、博士伦以及德国的蔡司,他们共占据市场的60%-70%份额,其中爱尔康一家独大,约占市场份额的30%。
 (图:2019年我国人工晶体各梯队市场份额)
(图:2019年我国人工晶体各梯队市场份额)
(图片来源:前瞻研究院 思宇医械观察整理)
国内而言,昊海生科是人工晶状体市场的重要参与者,约占国内市场的30%,2015年通过并购河南宇宙和珠海艾格等公司,昊海生科进入了人工晶体赛道,目前已经成为公司重要的盈利来源。其2019年报显示营收比例的44%来自于眼科产业。
此外,国内爱博诺德也是国内的IOL企业代表,爱博诺德公司在2014年发布的第一款核心产品“普诺明A1-UV”打破了国际高端软性可折叠人工晶状体的垄断,其价格比国际同类产品低30%至50%。据其2019年招股书,约占中国7%的IOL市场,但主要集中于中高端产品。2019年IOL全球市场份额为38亿美元,其仅占0.75%,相比于国内逊色不少。
高值耗材全国集采对人工晶体的未来影响 11月5日,售价约为13,000元的冠状动脉支架由于集采降价95%的新闻引起了轰动。由于这次采购的产品预计将占据公立医院的70%使用量,没有进入名单的企业将面临的业绩巨大下滑已可被预期。(详见:往期文章) 参考冠脉支架带量采购过程,人工晶体很有可能在未来进入全国集采环节。早在5月7日,国家医保局发布通知,要求省级平台报送冠脉支架、冠脉球囊、人工晶体三类产品2019年采购价格和采购量。当带量集采扩大范围,政策成熟之后,或许明年就将轮到IOL行业。
国产支架的市场占有率和产品系列相比于国外,均有很大提升空间,一旦实行IOL带量采购,是否会成为像昊海生科、爱博诺德等国内企业的弯道超车点还未明晰,可以有所期待。
结语 近二三十年来,鉴于患者需求改变,我国白内障在手术技术、器械、仪器设备、人工晶状体等方面都得到了快速发展,中国拥有世界上最大的白内障患者群体。人工晶状体作为白内障治疗不可少的光学器件,在我国仍属于蓝海市场,产品创新和政策动向都将成为影响行业的重要关注点,同时作为投资方向,人工晶体赛道也值得关注。
文章及图片来源:医用耗材研发


