






质量管理
-
2020.07.17
质量为先:测试、计量、检验和校准设备及用品抢先看!
本期展品预览小编为您带来的是将在2020Medtec中国展中亮相的测试、计量、检验和校准设备及用品供应商

阅读更多 -
2020.05.09
医疗器械的血液相容性评估
溶血和血栓形成的风险以及制造和加工参数的变化带来的影响?

阅读更多 -
2020.03.31
如何避免医疗器械包装穿孔?
造成包装穿孔后果极其严重,常见原因有三个:弯折开裂、磨损和直线穿刺。

阅读更多 -
2020.02.27
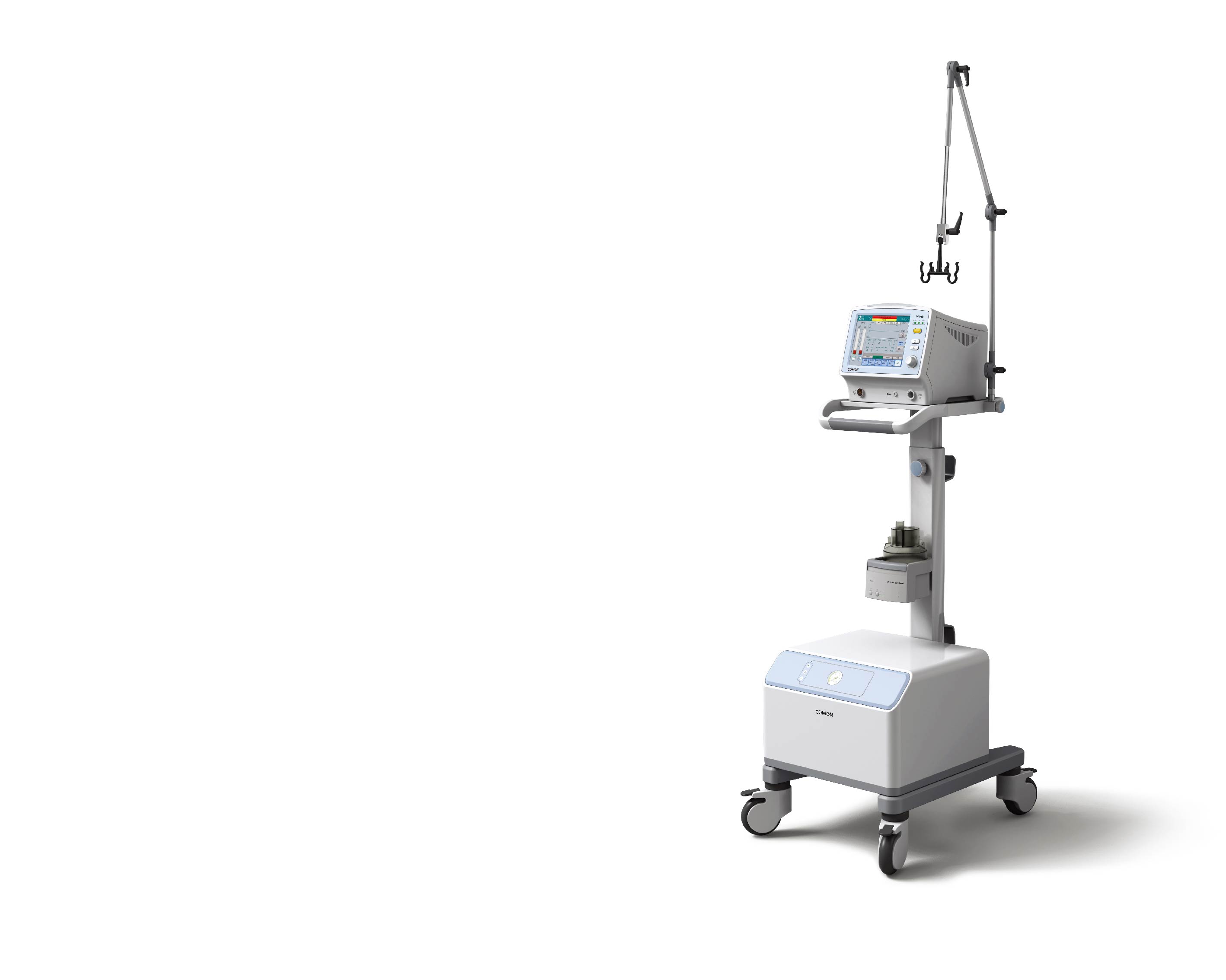
呼吸机的检测要点
呼吸机是肺部出现病变导致自主呼吸困难的患者的常用医疗设备,呼吸机可完全脱离呼吸中枢的调节和控制,人为地产生呼吸动作,以满足人体呼吸功能的需要。
阅读更多 -
2020.01.13
无菌医疗器械生产企业EO灭菌控制常见缺陷及监管要点分析
近年来,无菌医疗器械生产企业成为医疗器械GMP飞行检查的关注重点。生产企业从业人员可通过了解上述共性缺陷,掌握监管要点,在日常工作中配合法规标准的学习进行自查自纠,快速提升企业生产质量管理水平。政府监管人员则可通过系统了解法规标准的判定准则,掌握灭菌控制常见的共性缺陷特征,统一判定尺度,提升检查效率。
阅读更多 -
2019.11.15
确保患者安全,微粒至关重要
全世界每天都有成千上万的人要进行手术。无论是简单的微创手术,还是更复杂的骨科和心血管手术等,患者的健康和安全始终是最关键的问题,当将医疗器械植入体内时尤其如此。正是出于这个原因,所有三类医疗器械,包括用于心血管外科手术的器械,都要进行微粒检测,因为一旦植入体内,器械上残留的任何污染物都可能导致包括闭塞或感染在内的并发症。

阅读更多 -
2019.11.12
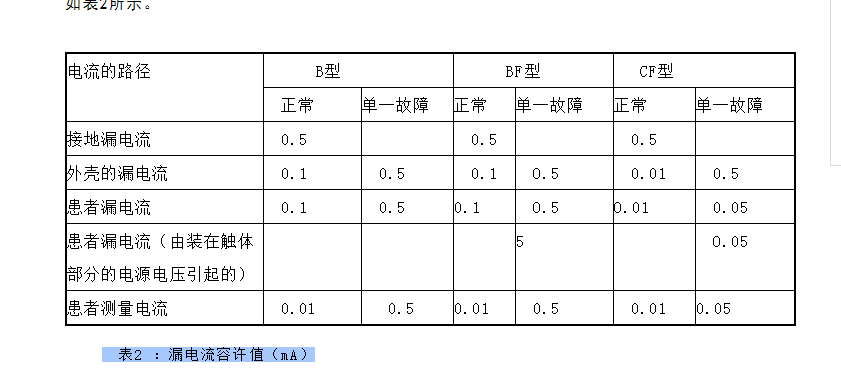
医疗器械的电气安全措施及其分类
医学仪器的电气安全主要是指仪器在使用时防止电击的性能,它是医学仪器安全性的重要组成部分。
阅读更多 -
2019.09.20
质量管理及质量控制宝典
APQP(Advanced Product Quality Planning)即产品质量先期策划,是一种结构化的方法,用来确定和制定确保某产品使顾客满意所需的步骤。
阅读更多 -
2019.09.18
Quality Expo2019-测试、计量、检验和校准设备及用品推荐(2)
本期Medtec中国展展品预览为大家带来的是测试、计量、检验和校准设备及用品品类,汇聚了来自全球的顶级的供应商,产品包括Sprint mD、Sprint iQ、桡动脉压缩带气密检测设备、体外血液循坏系统、侵入式医疗器械的气密检测仪器、针对血管介入开发的系统性的模拟器、先步机器人真空采血管、医疗产品测试一站式解决方案、X射线透工业CT-DeskTom、AnTestin平台、Minitab软件、HDS-PRO系列全自动扫描显微镜……先跟小编来提前看一部分企业和产品介绍吧。

阅读更多 -
2019.08.08
医疗器械生产质量管理规范核查常见问题与浅析
根据《国家食品药品监督管理总局发布医疗器械注册管理办法》(国家食品药品监督管理总局令第4号)的规定,境内第二类、第三类医疗器械注册质量管理体系核查,由省、自治区、直辖市食品药品监督管理部门开展,本文主要就境内二类医疗器械的质量管理规范核查过程中出现的常见问题进行分析研究。
阅读更多


